Cao Duc Tuan, Huong Doan Thi Mai, Pham Van Cuong, Minh Le Thi Hong, Chae Jung-Woo, Yun Hwi-Yeol, Na Min-Kyun, Kim Young-Ho, Pham Minh Quan, Nguyen Van Hung
Hai Phong University of Medicine and Pharmacy Haiphong Vietnam
Institute of Marine Biochemistry, Vietnam Academy of Science and Technology Hanoi Vietnam.
RSC Adv. 2021 Jun 4;11(33):20173-20179. doi: 10.1039/d1ra01855h. eCollection 2021 Jun 3.
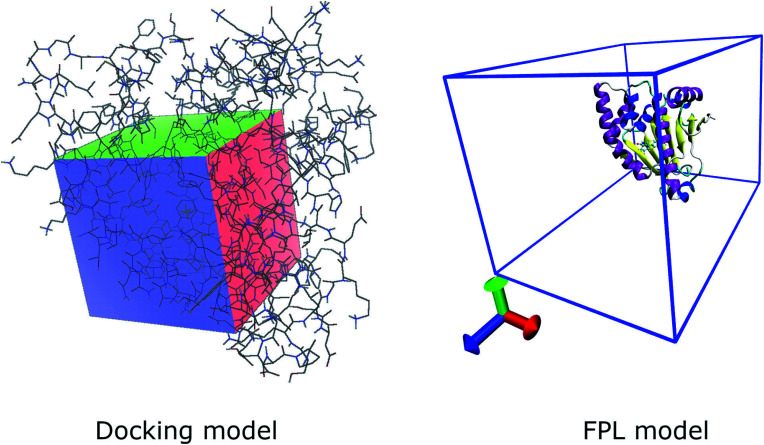
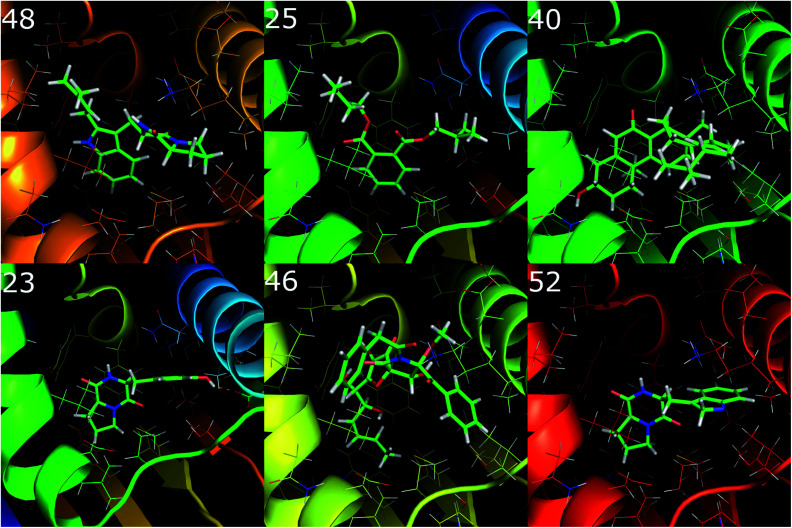
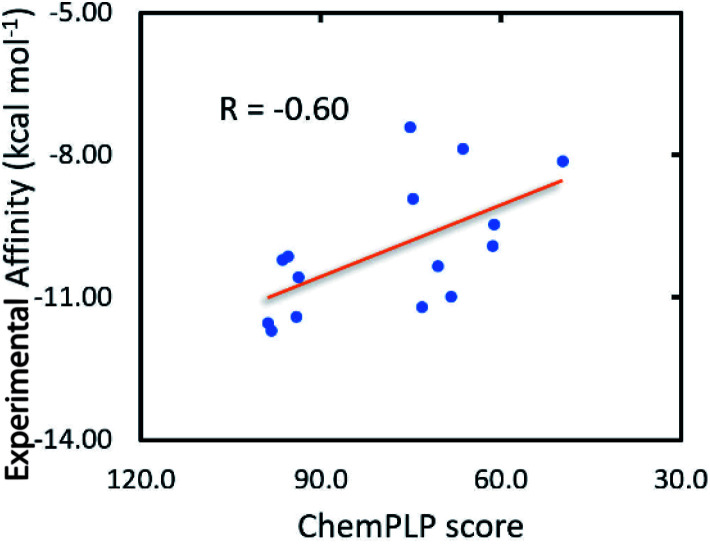
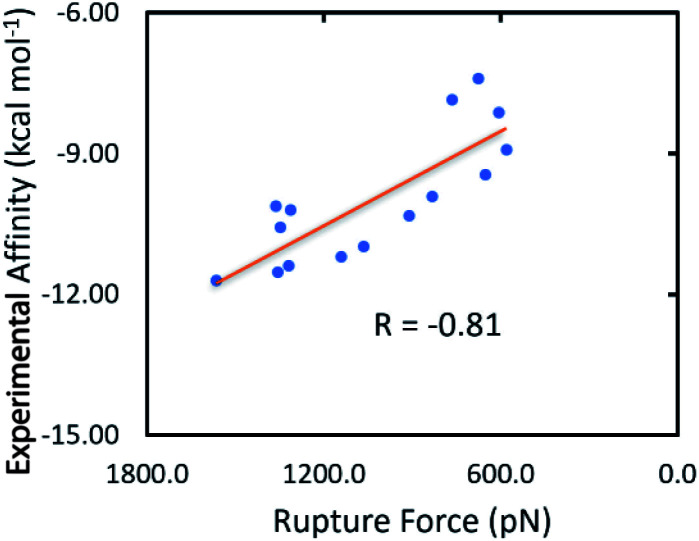
Heat shock protein 90 (Hsp90) is one of the most potential targets in cancer therapy. We have demonstrated using a combination of molecular docking and fast pulling of ligand (FPL) simulations that marine fungi derivatives can be possible inhibitors, preventing the biological activity of Hsp90. The computational approaches were validated and compared with previous experiments. Based on the benchmark of available inhibitors of Hsp90, the GOLD docking package using the ChemPLP scoring function was found to be superior over both Autodock Vina and Autodock4 in the preliminary estimation of the ligand-binding affinity and binding pose with the Pearson correlation, = -0.62. Moreover, FPL calculations were also indicated as a suitable approach to refine docking simulations with a correlation coefficient with the experimental data of = -0.81. Therefore, the binding affinity of marine fungi derivatives to Hsp90 was evaluated. Docking and FPL calculations suggest that five compounds including 23, 40, 46, 48, and 52 are highly potent inhibitors for Hsp90. The obtained results enhance cancer therapy research.
热休克蛋白90(Hsp90)是癌症治疗中最具潜力的靶点之一。我们通过分子对接和配体快速拉动(FPL)模拟相结合的方法证明,海洋真菌衍生物可能是抑制剂,可阻止Hsp90的生物活性。这些计算方法经过了验证,并与之前的实验进行了比较。基于Hsp90现有抑制剂的基准,发现在配体结合亲和力和结合构象的初步估计中,使用ChemPLP评分函数的GOLD对接软件包优于Autodock Vina和Autodock4,皮尔逊相关系数为 = -0.62。此外,FPL计算也被表明是一种合适的方法来优化对接模拟,与实验数据的相关系数为 = -0.81。因此,评估了海洋真菌衍生物与Hsp90的结合亲和力。对接和FPL计算表明,包括23、40、46、48和52在内的五种化合物是Hsp90的高效抑制剂。所得结果推动了癌症治疗研究。