Ngo Son Tung, Mai Binh Khanh, Derreumaux Philippe, Vu Van V
Laboratory of Theoretical and Computational Biophysics, Ton Duc Thang University Ho Chi Minh City Vietnam
Faculty of Applied Sciences, Ton Duc Thang University Ho Chi Minh City Vietnam.
RSC Adv. 2019 Apr 23;9(22):12455-12461. doi: 10.1039/c9ra01177c. eCollection 2019 Apr 17.
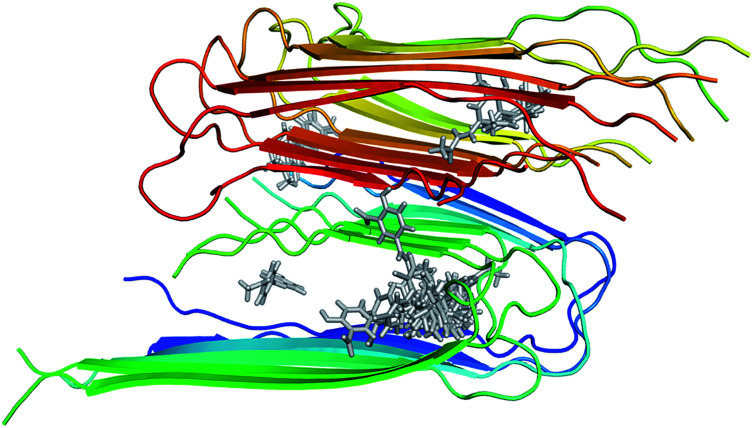
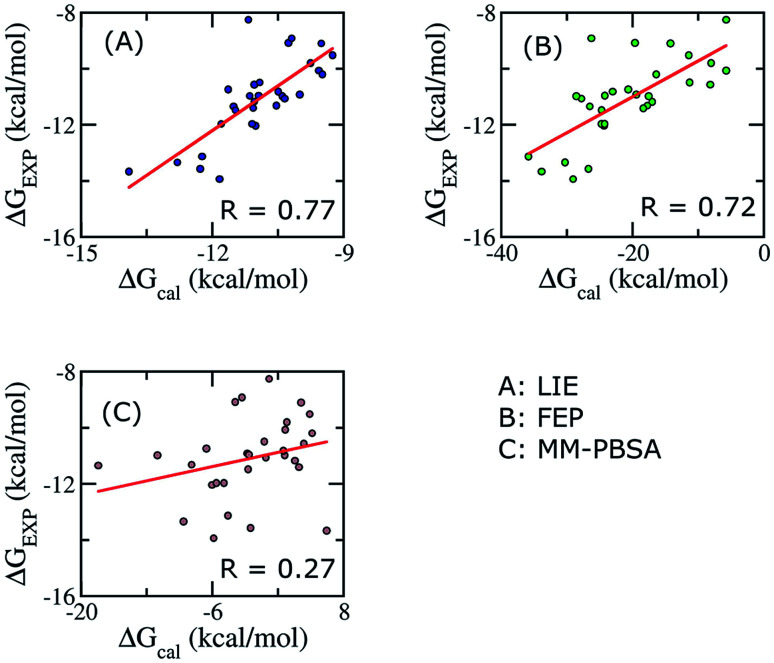
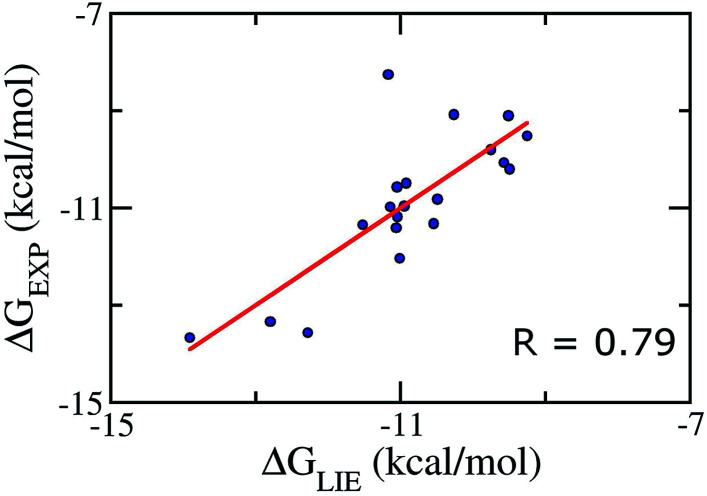
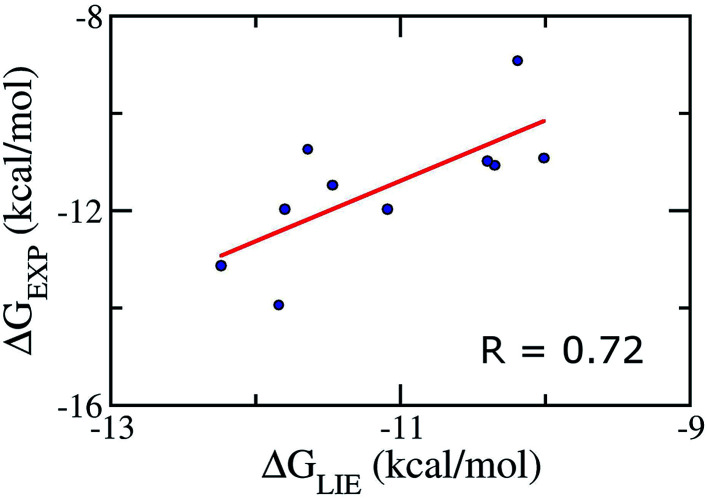
The search for efficient inhibitors targeting Aβ oligomers and fibrils is an important issue in Alzheimer's disease treatment. As a consequence, an accurate and computationally cheap approach to estimate the binding affinity for many ligands interacting with Aβ peptides is very important. Here, the calculated binding free energies of 30 ligands interacting with 12Aβ peptides using the linear interaction energy (LIE) approach are found to be in good correlation with experimental data ( = 0.79). The binding affinities of these complexes are also calculated by using free energy perturbation (FEP) and molecular mechanic/Poisson-Boltzmann surface area (MM/PBSA) methods. The time-consuming FEP method provides results with similar correlation ( = 0.72), whereas MM/PBSA calculations show very low correlation with experimental data ( = 0.27). In all complexes, van der Waals interactions contribute much more than electrostatic interactions. The LIE model, which is much less time-consuming than both the FEP and MM/PBSA methods, opens the door to accurate and rapid affinity prediction of ligands with Aβ peptides and the design of new ligands.
寻找针对淀粉样β蛋白(Aβ)寡聚体和原纤维的有效抑制剂是阿尔茨海默病治疗中的一个重要问题。因此,一种准确且计算成本低的方法来估计许多与Aβ肽相互作用的配体的结合亲和力非常重要。在此,使用线性相互作用能(LIE)方法计算的30种配体与12种Aβ肽相互作用的结合自由能与实验数据具有良好的相关性( = 0.79)。这些复合物的结合亲和力也通过自由能微扰(FEP)和分子力学/泊松-玻尔兹曼表面积(MM/PBSA)方法进行计算。耗时的FEP方法提供了具有相似相关性的结果( = 0.72),而MM/PBSA计算显示与实验数据的相关性非常低( = 0.27)。在所有复合物中,范德华相互作用的贡献远大于静电相互作用。LIE模型比FEP和MM/PBSA方法耗时少得多,为准确快速预测配体与Aβ肽的亲和力以及设计新配体打开了大门。