Yao Yuanshan, Li Zheng, Gao Wen
Department of Thoracic Surgery, Shanghai Key Laboratory of Clinical Geriatric Medicine, HuaDong Hospital Affiliated to Fudan University, Shanghai, China.
Front Genet. 2022 Apr 11;13:855789. doi: 10.3389/fgene.2022.855789. eCollection 2022.
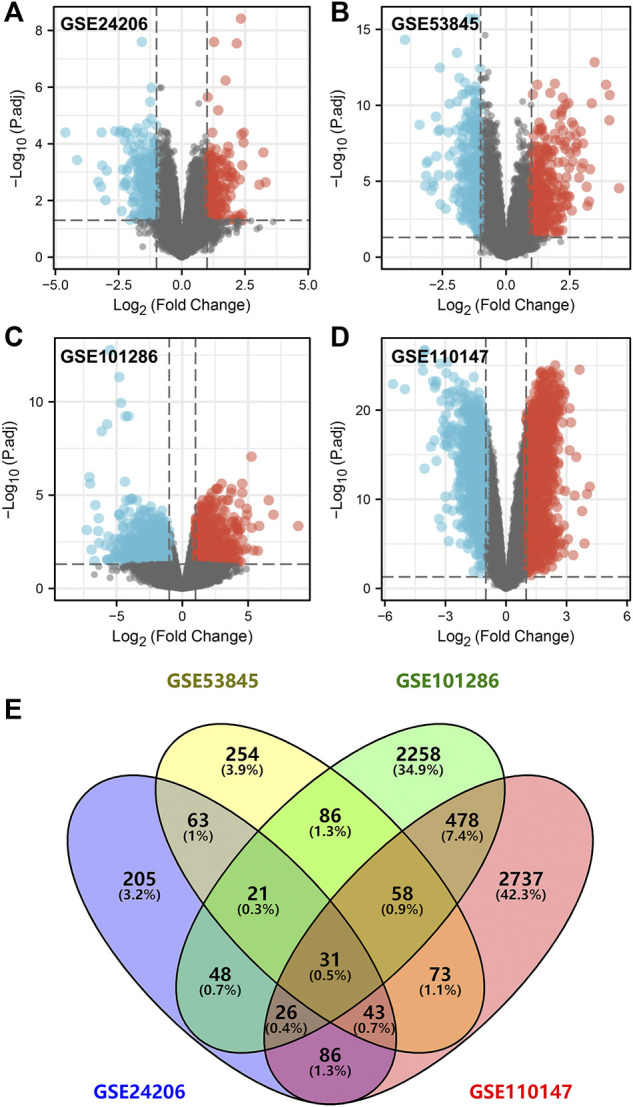
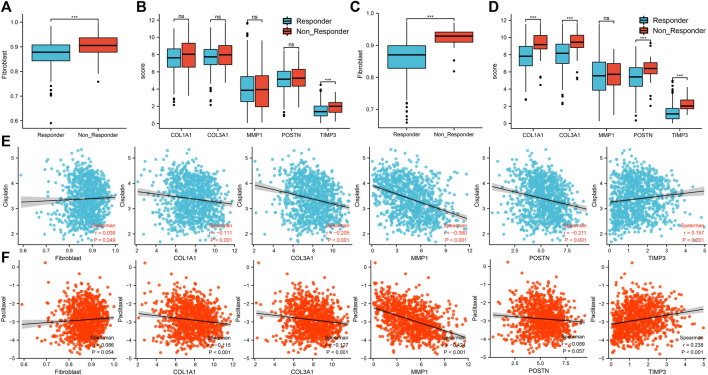
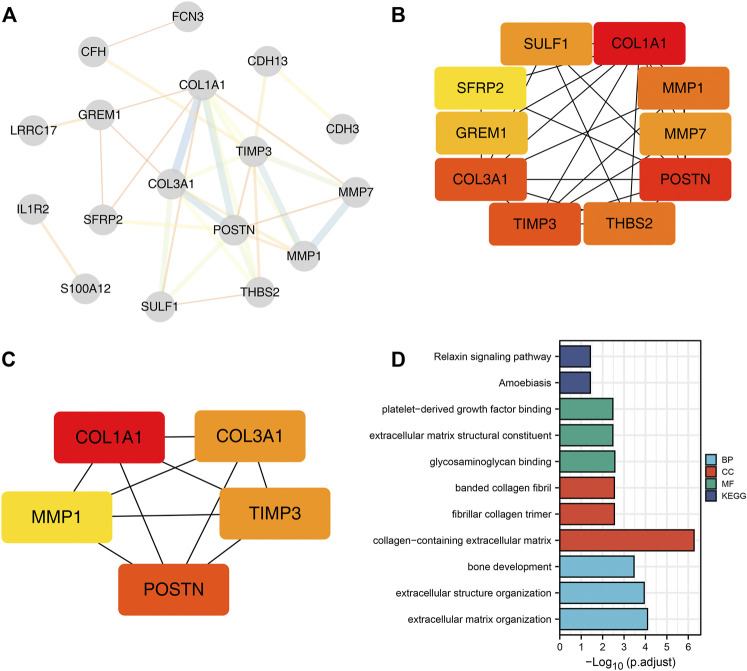
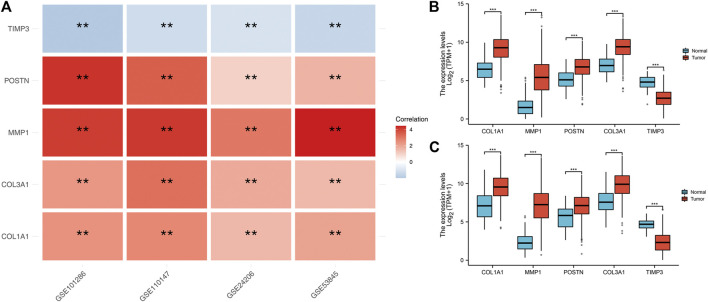
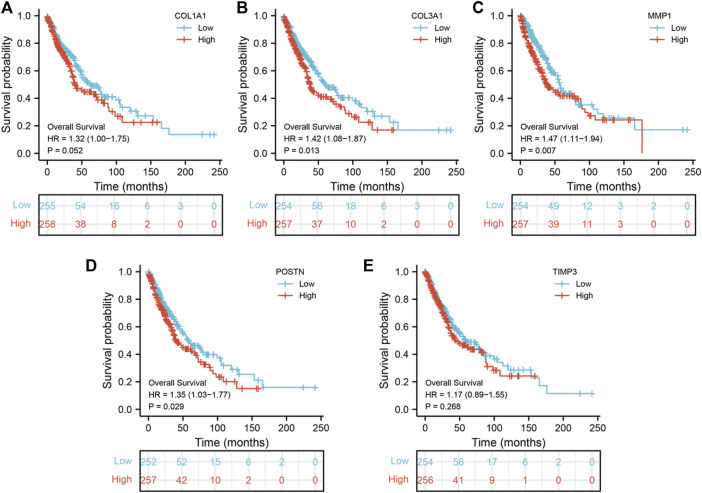
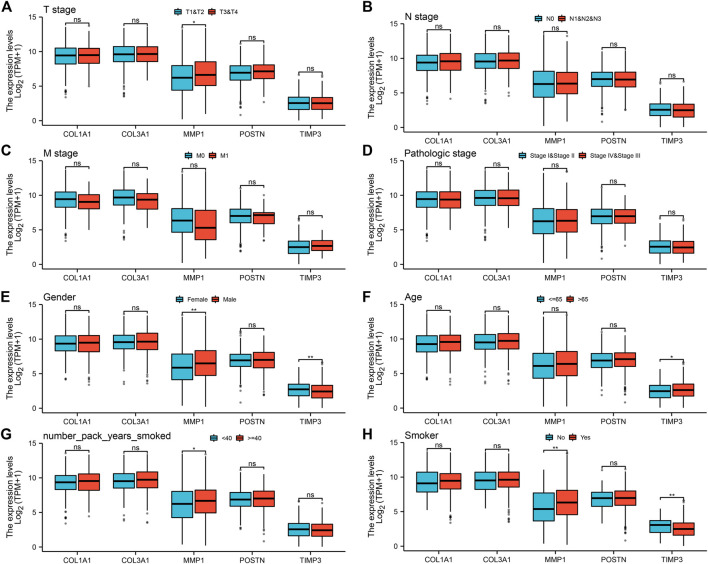
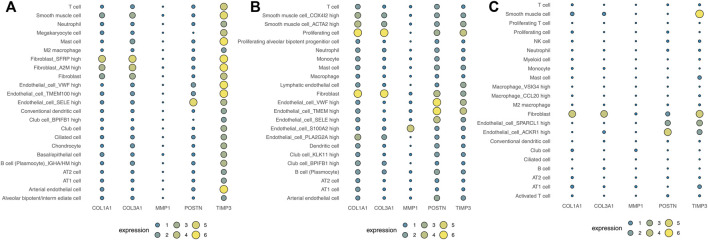
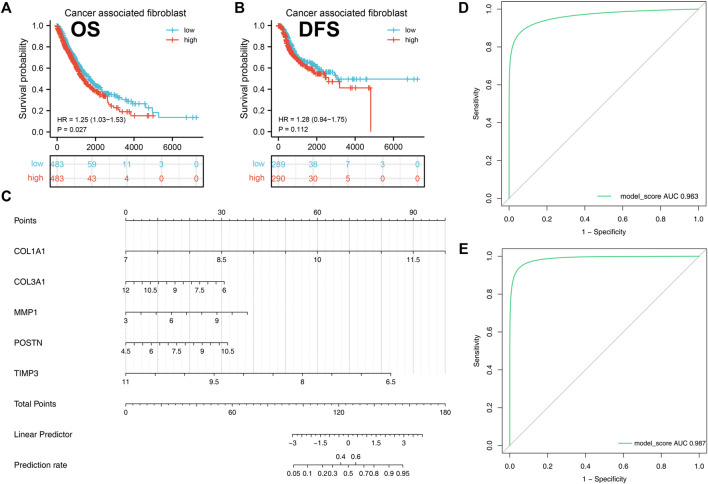
Lung cancer is the most common comorbidity of idiopathic pulmonary fibrosis. Thus there is an urgent need for the research of IPF and carcinogenesis The objective of this study was to explore hub genes which are common in pulmonary fibrosis and lung cancer progression through bioinformatic analysis. All the analysis was performed in R software. Differentially expressed genes (DEGs) were explored by comparing gene expression profiles between IPF tissues and healthy lung tissues from GSE24206, GSE53845, GSE101286 and GSE110147 datasets. Venn Diagram analysis was used to identify the overlapping genes, while GO and KEGG pathway enrichment analysis were used to explore the biological functions of the DEGs using clusterprofiler package. Hub genes were identified by analyzing protein-protein interaction networks using Cytoscape software. Nomogram was constructed using the rms package. Tumor immune dysfunction and exclusion (TIDE) and Genomics of Drug Sensitivity in Cancer (GDSC) analysis was used to quantify the immunotherapy and chemotherapy sensitivity of non-small cell lung cancer (NSCLC) patients. and were identified as the top five hub genes. The five hub genes were used to construct a diagnostic nomogram that was validated in another IPF dataset. Since the hub genes were also associated with lung cancer progression, we found that the nomogram also had diagnostic value in NSCLC patients. These five genes achieved a statistically difference of overall survival in NSCLC patients ( < 0.05). The expression of the five hub genes was mostly enriched in fibroblasts. Fibroblasts and the hub genes also showed significant ability to predict the susceptibility of NSCLC patients to chemotherapy and immunotherapy. We identified five hub genes as potential biomarkers of IPF and NSCLC progression. This finding may give insight into the underlying molecular mechanisms of IPF and lung cancer progression and provides potential targets for developing new therapeutic agents for IPF patients.
肺癌是特发性肺纤维化最常见的合并症。因此,迫切需要对特发性肺纤维化和致癌作用进行研究。本研究的目的是通过生物信息学分析探索在肺纤维化和肺癌进展中常见的关键基因。所有分析均在R软件中进行。通过比较来自GSE24206、GSE53845、GSE101286和GSE110147数据集的特发性肺纤维化组织与健康肺组织之间的基因表达谱,探索差异表达基因(DEGs)。使用维恩图分析来识别重叠基因,同时使用clusterprofiler包通过基因本体(GO)和京都基因与基因组百科全书(KEGG)通路富集分析来探索DEGs的生物学功能。使用Cytoscape软件分析蛋白质-蛋白质相互作用网络来识别关键基因。使用rms包构建列线图。使用肿瘤免疫功能障碍与排除(TIDE)和癌症药物敏感性基因组学(GDSC)分析来量化非小细胞肺癌(NSCLC)患者的免疫治疗和化疗敏感性。 和 被确定为前五个关键基因。这五个关键基因被用于构建一个诊断列线图,并在另一个特发性肺纤维化数据集中进行了验证。由于这些关键基因也与肺癌进展相关,我们发现该列线图对NSCLC患者也具有诊断价值。这五个基因在NSCLC患者的总生存期上具有统计学差异(<0.05)。这五个关键基因的表达大多在成纤维细胞中富集。成纤维细胞和关键基因也显示出显著的能力来预测NSCLC患者对化疗和免疫治疗的易感性。我们确定了五个关键基因作为特发性肺纤维化和NSCLC进展的潜在生物标志物。这一发现可能有助于深入了解特发性肺纤维化和肺癌进展的潜在分子机制,并为开发针对特发性肺纤维化患者的新型治疗药物提供潜在靶点。