Niu Yuzhen, Yao Xiaojun, Ji Hongfang
Shandong Provincial Research Center for Bioinformatic Engineering and Technique, College of Life Sciences, Shandong University of Technology Zibo 255049 China
State Key Laboratory of Applied Organic Chemistry, Department of Chemistry, Lanzhou University Lanzhou 730000 China.
RSC Adv. 2019 Apr 23;9(22):12441-12454. doi: 10.1039/c9ra01657k. eCollection 2019 Apr 17.
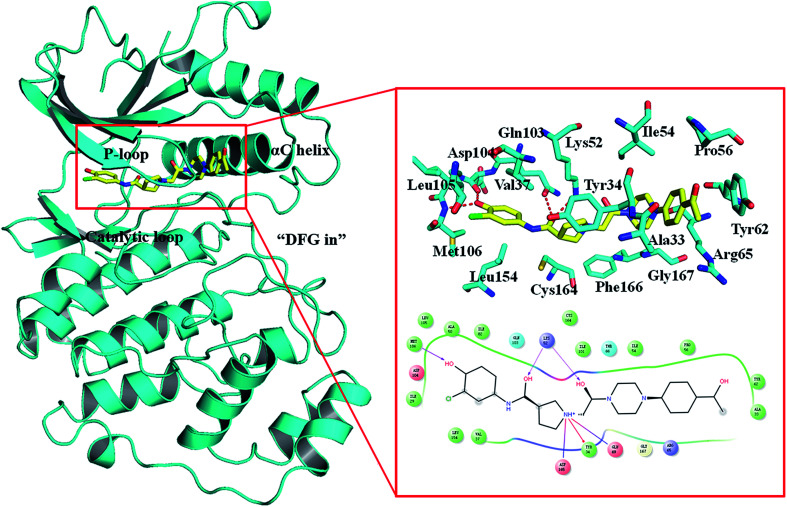
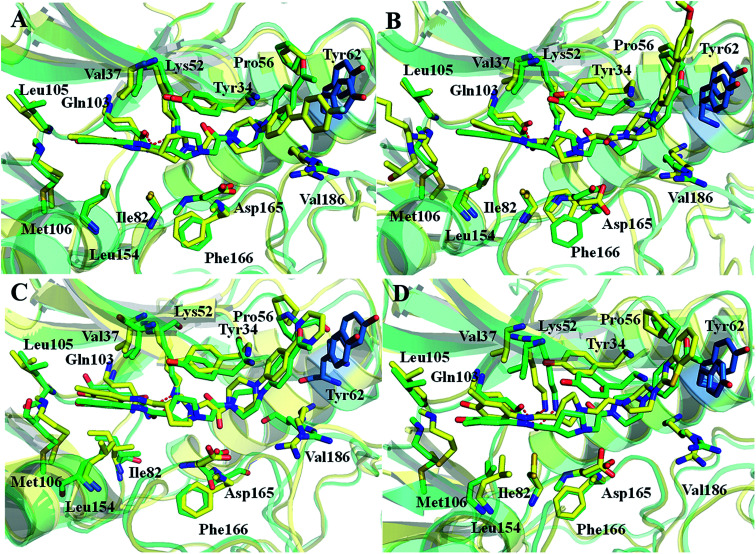
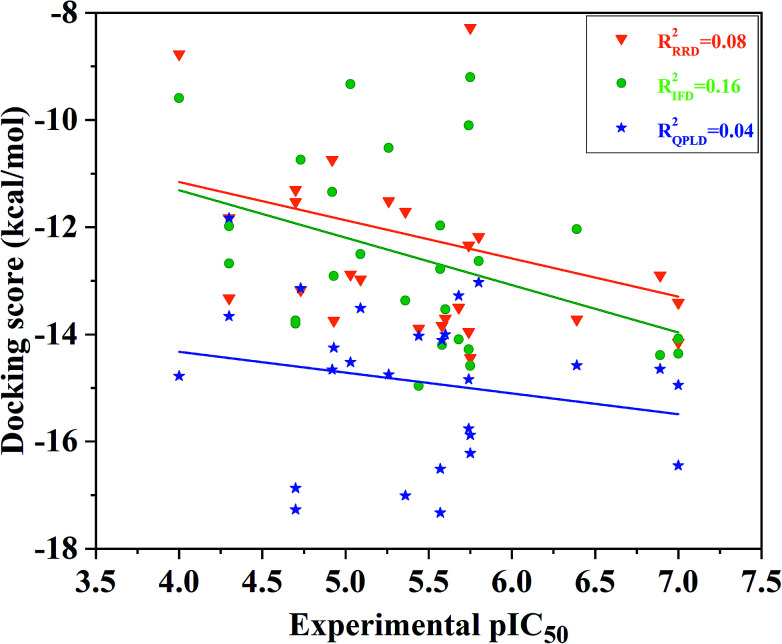
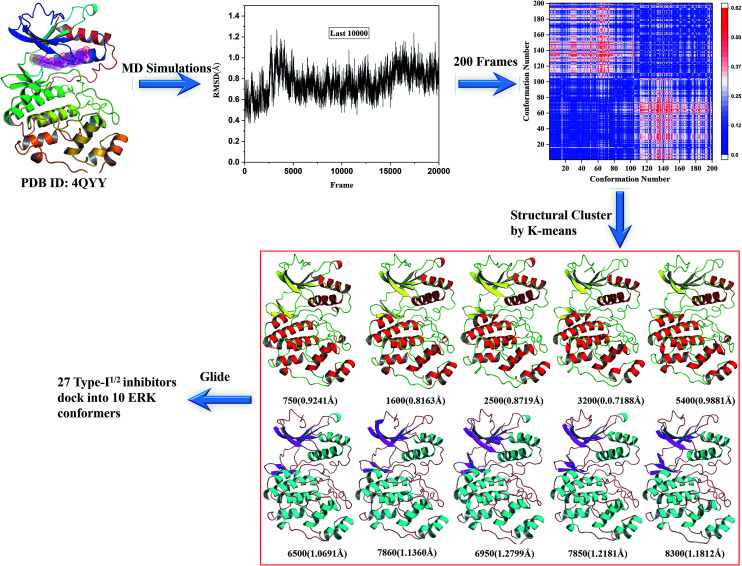
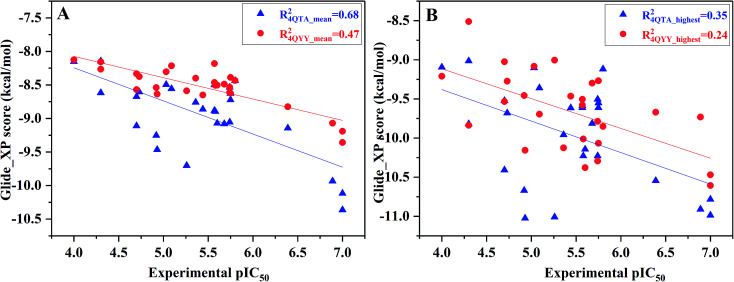
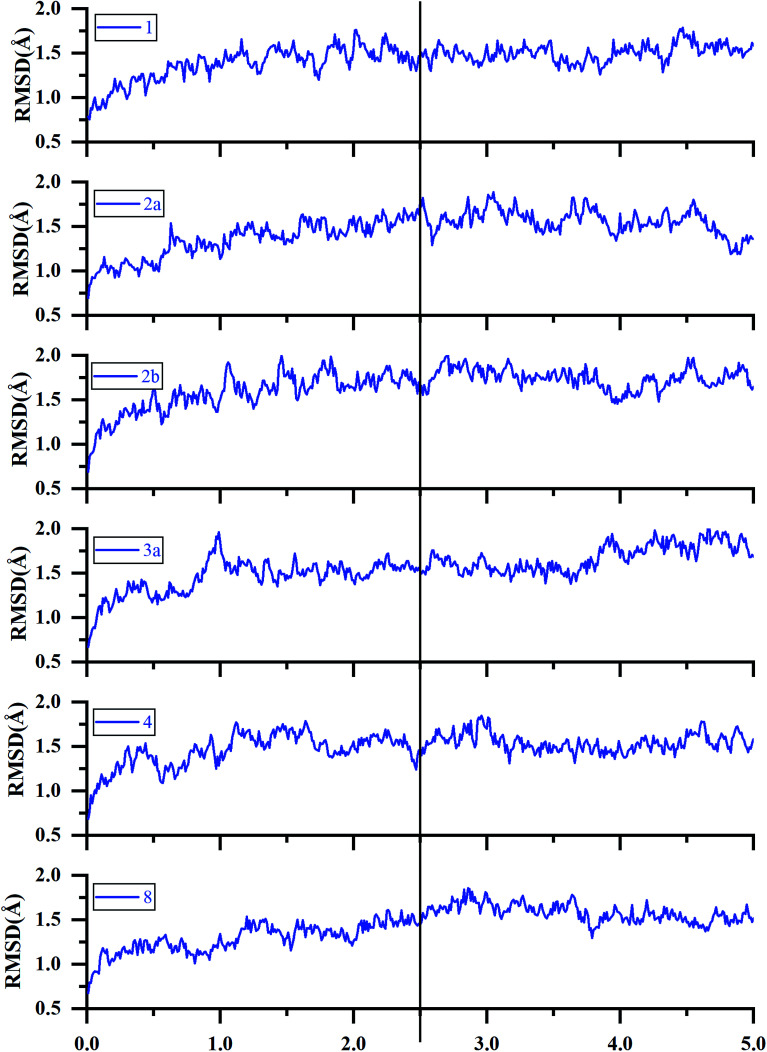
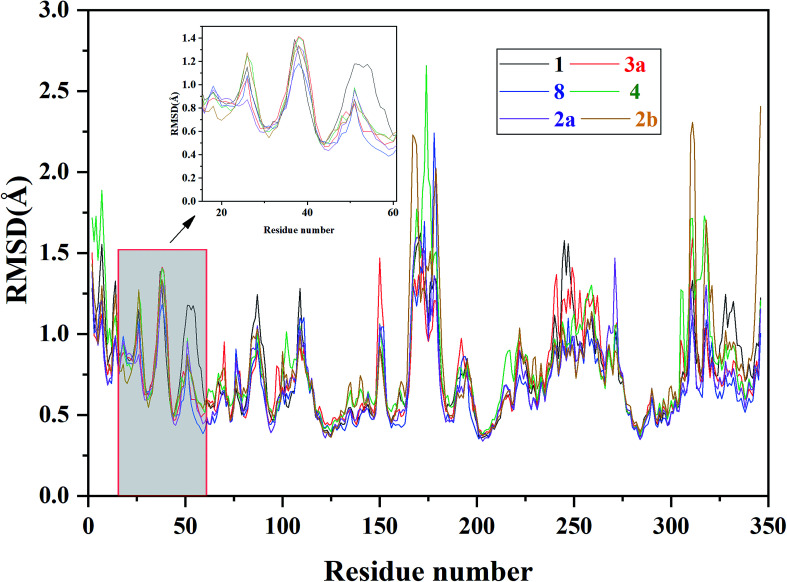
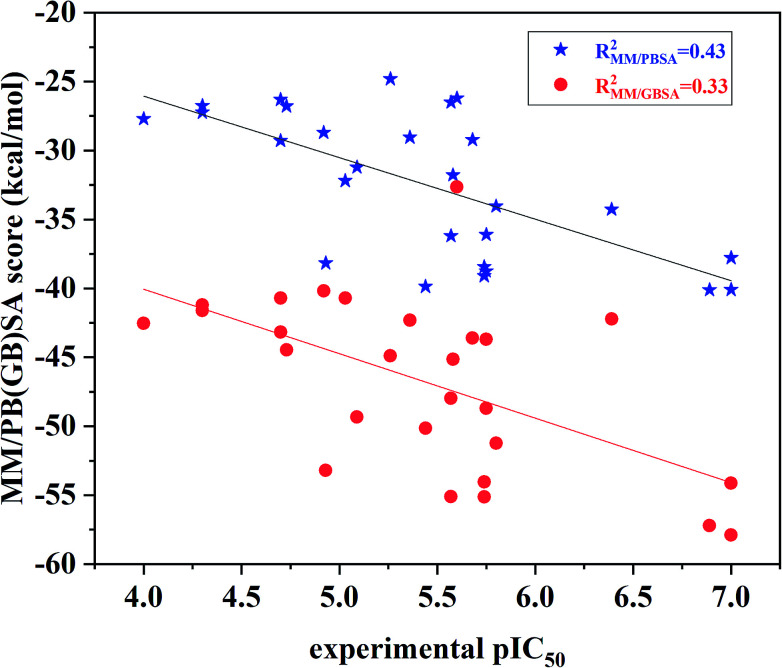
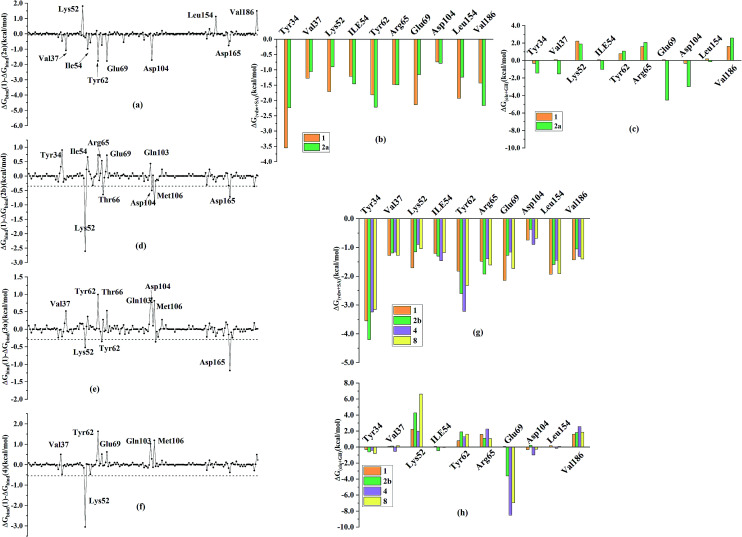
Extracellular-regulated kinase (ERK2) has been regarded as an essential target for various cancers, especially melanoma. Recently, pyrrolidine piperidine derivatives were reported as Type I inhibitors of ERK2, which occupy both the ATP binding pocket and the allosteric pocket. Due to the dynamic behavior of ERK2 upon the binding of Type I inhibitors, it is difficult to predict the binding structures and relative binding potencies of these inhibitors with ERK2 accurately. In this work, the binding mechanism of pyrrolidine piperidines was discussed by using different simulation techniques, including molecular docking, ensemble docking based on multiple receptor conformation, molecular dynamics simulations and free energy calculations. Our computational results show that the traditional docking method cannot predict the relative binding ability of the studied inhibitors with high accuracy, but incorporating ERK2 protein flexibility into docking is an effective method to improve the prediction accuracy. It is worth noting that the binding free energies predicted by MM/GBSA or MM/PBSA based on the MD simulations for the docked poses have the highest correlation with the experimental data, which highlights the importance of protein flexibility for accurately predicting the binding ability of Type I inhibitors of ERK2. In addition, the comprehensive analysis of several representative inhibitors indicates that hydrogen bonds and hydrophobic interactions are of significance for improving the binding affinities of the inhibitors. We hope this work will provide valuable information for further design of novel and efficient Type I ERK2 inhibitors.
细胞外调节激酶(ERK2)一直被视为各种癌症,尤其是黑色素瘤的重要靶点。最近,吡咯烷哌啶衍生物被报道为ERK2的I型抑制剂,它们占据了ATP结合口袋和别构口袋。由于I型抑制剂结合后ERK2的动态行为,很难准确预测这些抑制剂与ERK2的结合结构和相对结合能力。在这项工作中,通过使用不同的模拟技术,包括分子对接、基于多受体构象的 ensemble对接、分子动力学模拟和自由能计算,讨论了吡咯烷哌啶的结合机制。我们的计算结果表明,传统的对接方法不能高精度地预测所研究抑制剂的相对结合能力,但将ERK2蛋白的灵活性纳入对接是提高预测准确性的有效方法。值得注意的是,基于对接姿势的MD模拟通过MM/GBSA或MM/PBSA预测的结合自由能与实验数据具有最高的相关性,这突出了蛋白灵活性对于准确预测ERK2的I型抑制剂结合能力的重要性。此外,对几种代表性抑制剂的综合分析表明,氢键和疏水相互作用对于提高抑制剂的结合亲和力具有重要意义。我们希望这项工作将为进一步设计新型高效的ERK2的I型抑制剂提供有价值的信息。