Liu Bangquan, Zhai Jiabao, Wang Wanyu, Liu Tianyu, Liu Chang, Zhu Xiaojie, Wang Qi, Tian Wenjing, Zhang Fubin
Department of Epidemiology, College of Public Health, Harbin Medical University, Harbin, China.
Department of Gynecological Oncology, Harbin Medical University Cancer Hospital, Harbin, China.
Front Mol Biosci. 2022 Apr 19;9:872932. doi: 10.3389/fmolb.2022.872932. eCollection 2022.
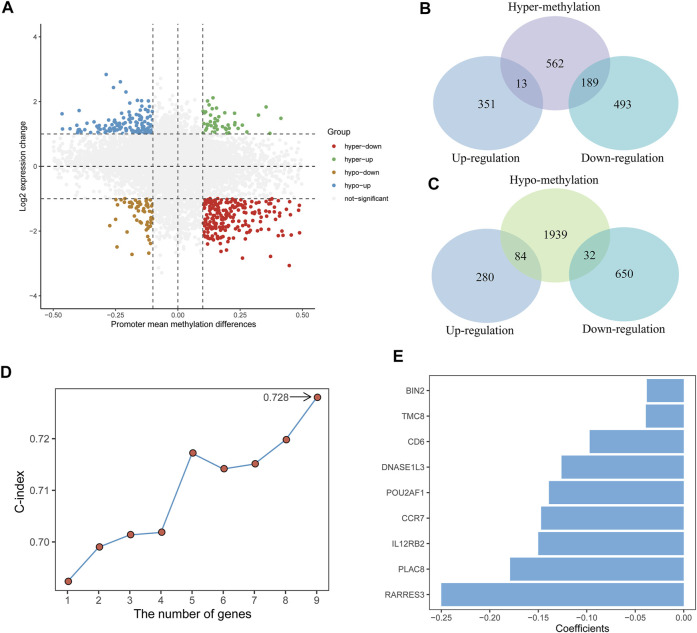
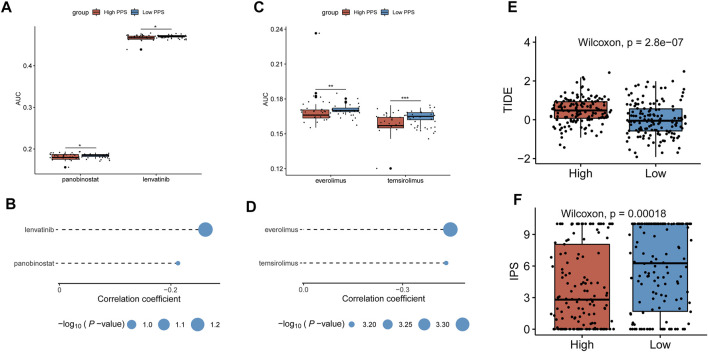
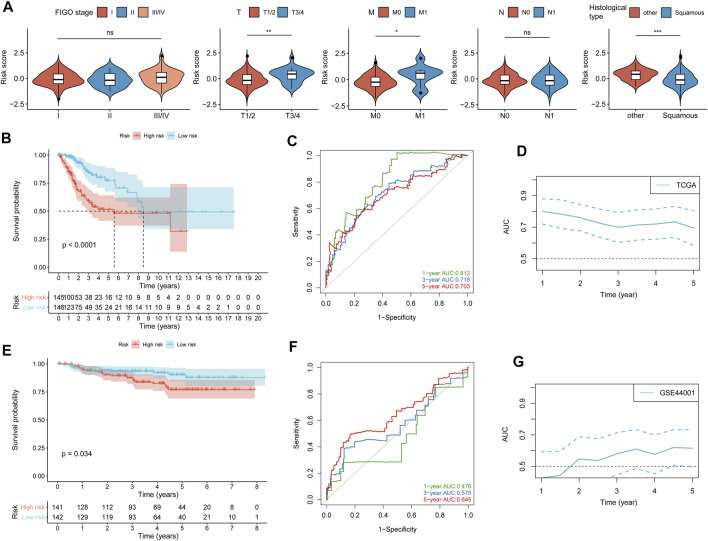
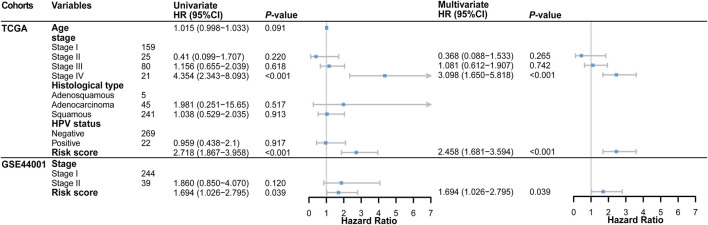
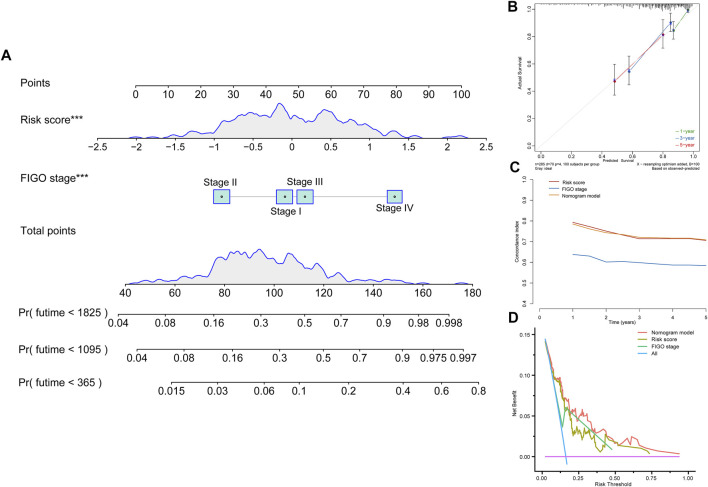
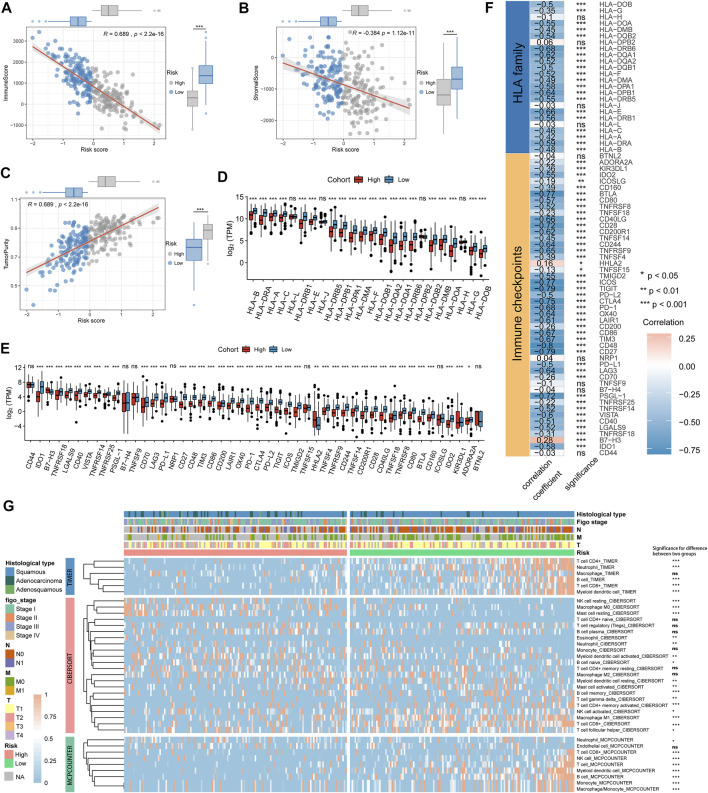
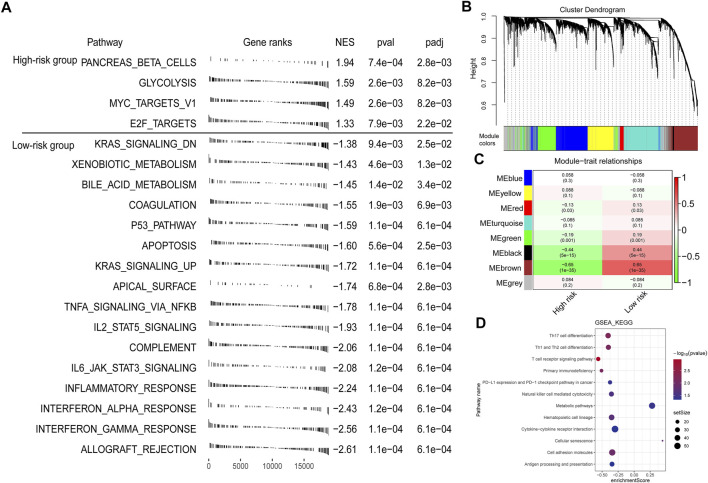
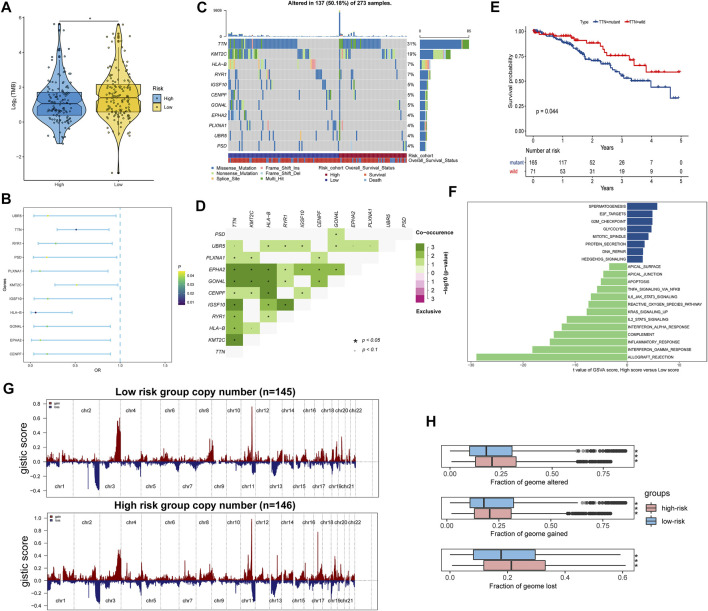
Tumor microenvironment (TME) has been reported to have a strong association with tumor progression and therapeutic outcome, and epigenetic modifications such as DNA methylation can affect TMB and play an indispensable role in tumorigenesis. However, the potential mechanisms of TME and DNA methylation remain unclear in cervical cancer (CC). The immune and stromal scores of TME were generated by the ESTIMATE algorithm for CC patients in The Cancer Genome Atlas (TCGA) database. The TME and DNA methylation-related genes were identified by the integrative analysis of DNA promoter methylation and gene expression. The least absolute shrinkage and selection operator (LASSO) Cox regression was performed 1,000 times to further identify a nine-gene TME and DNA methylation-related prognostic signature. The signature was further validated in Gene Expression Omnibus (GEO) dataset. Then, the identified signature was integrated with the Federation International of Gynecology and Obstetrics (FIGO) stage to establish a composite prognostic nomogram. CC patients with high immunity levels have better survival than those with low immunity levels. Both in the training and validation datasets, the risk score of the signature was an independent prognosis factor. The composite nomogram showed higher accuracy of prognosis and greater net benefits than the FIGO stage and the signature. The high-risk group had a significantly higher fraction of genome altered than the low-risk group. Eleven genes were significantly different in mutation frequencies between the high- and low-risk groups. Interestingly, patients with mutant had better overall survival (OS) than those with wild type. Patients in the low-risk group had significantly higher tumor mutational burden (TMB) than those in the high-risk group. Taken together, the results of TMB, immunophenoscore (IPS), and tumor immune dysfunction and exclusion (TIDE) score suggested that patients in the low-risk group may have greater immunotherapy benefits. Finally, four drugs (panobinostat, lenvatinib, everolimus, and temsirolimus) were found to have potential therapeutic implications for patients with a high-risk score. Our findings highlight that the TME and DNA methylation-related prognostic signature can accurately predict the prognosis of CC and may be important for stratified management of patients and precision targeted therapy.
据报道,肿瘤微环境(TME)与肿瘤进展和治疗结果密切相关,DNA甲基化等表观遗传修饰可影响肿瘤突变负荷(TMB)并在肿瘤发生中发挥不可或缺的作用。然而,在宫颈癌(CC)中,TME和DNA甲基化的潜在机制仍不清楚。利用癌症基因组图谱(TCGA)数据库中的ESTIMATE算法生成CC患者TME的免疫和基质评分。通过DNA启动子甲基化和基因表达的综合分析,鉴定出TME和DNA甲基化相关基因。进行1000次最小绝对收缩和选择算子(LASSO)Cox回归,以进一步鉴定出一个与TME和DNA甲基化相关的九基因预后特征。该特征在基因表达综合数据库(GEO)数据集中得到进一步验证。然后,将鉴定出的特征与国际妇产科联合会(FIGO)分期相结合,建立一个复合预后列线图。免疫水平高的CC患者比免疫水平低的患者生存更好。在训练和验证数据集中,该特征的风险评分都是一个独立的预后因素。与FIGO分期和该特征相比,复合列线图显示出更高的预后准确性和更大的净效益。高风险组的基因组改变比例显著高于低风险组。高风险组和低风险组之间有11个基因的突变频率存在显著差异。有趣的是,具有突变体的患者比野生型患者的总生存期(OS)更好。低风险组患者的肿瘤突变负荷(TMB)显著高于高风险组。综合来看,TMB、免疫表型评分(IPS)和肿瘤免疫功能障碍与排除(TIDE)评分的结果表明,低风险组患者可能从免疫治疗中获益更大。最后,发现四种药物(帕比司他、乐伐替尼、依维莫司和替西罗莫司)对高风险评分患者具有潜在的治疗意义。我们的研究结果表明,TME和DNA甲基化相关的预后特征可以准确预测CC的预后,可能对患者的分层管理和精准靶向治疗具有重要意义。