The Second Department of Cardiology, Department of Obstetrics and Gynecology, The Second People's Hospital of Guangdong Province, Guangzhou, 510310, China.
The First Dongguan Affiliated Hospital, Guangdong Medical University, Dongguan, 523710, Guangdong Province, China.
Orphanet J Rare Dis. 2022 May 7;17(1):183. doi: 10.1186/s13023-022-02348-z.
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is associated with ventricular arrhythmia, heart failure (HF), and sudden death. Thromboembolism is also an important and serious complication of ARVC/D. However, the etiology of ARVC/D and thromboembolism and their association with genetic mutations are unclear.
Genomic DNA samples of peripheral blood were conducted for whole-exome sequencing (WES) and Sanger sequencing in the ARVC/D family. Then, we performed bioinformatics analysis for genes susceptible to cardiomyopathies and arrhythmias. Further, we analyzed how the potential pathogenic mutations were affecting the hydrophobicity and phosphorylation of amino acids and their joint pathogenicity by ProtScale, NetPhos and ORVAL algorisms.
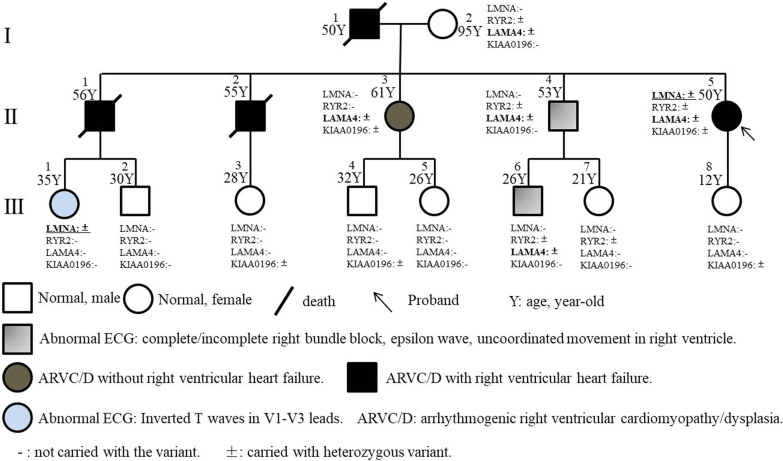
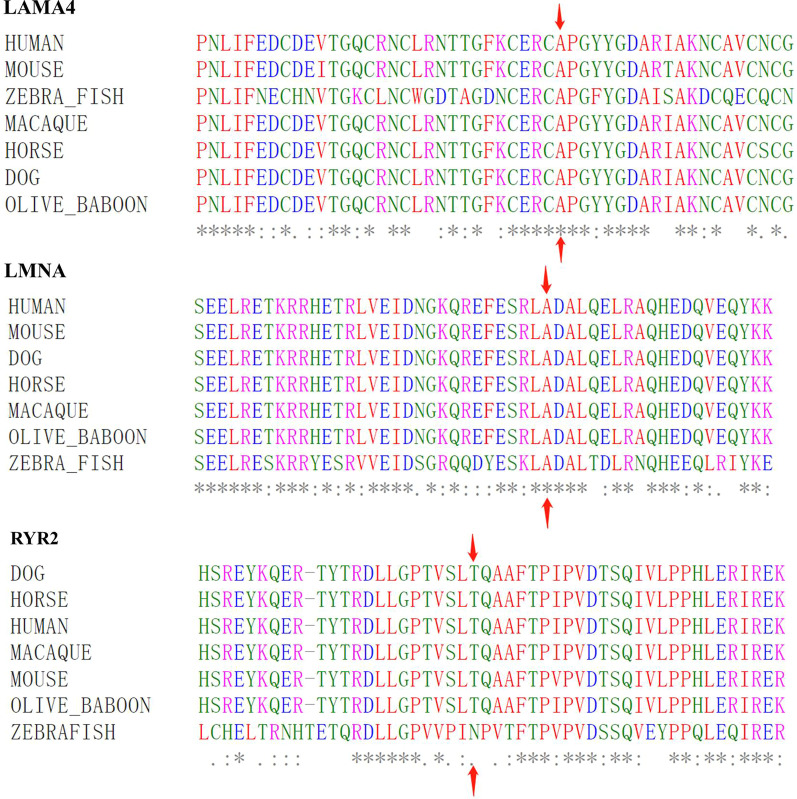
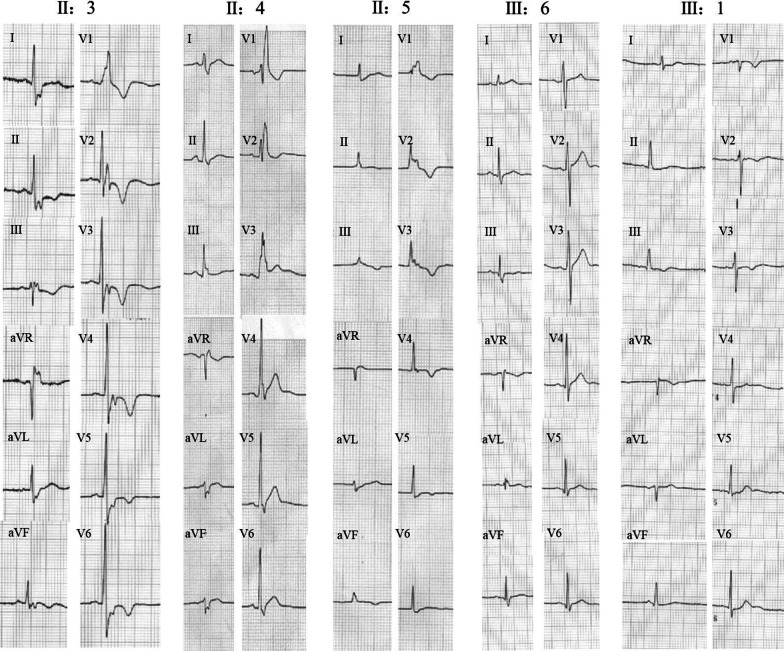
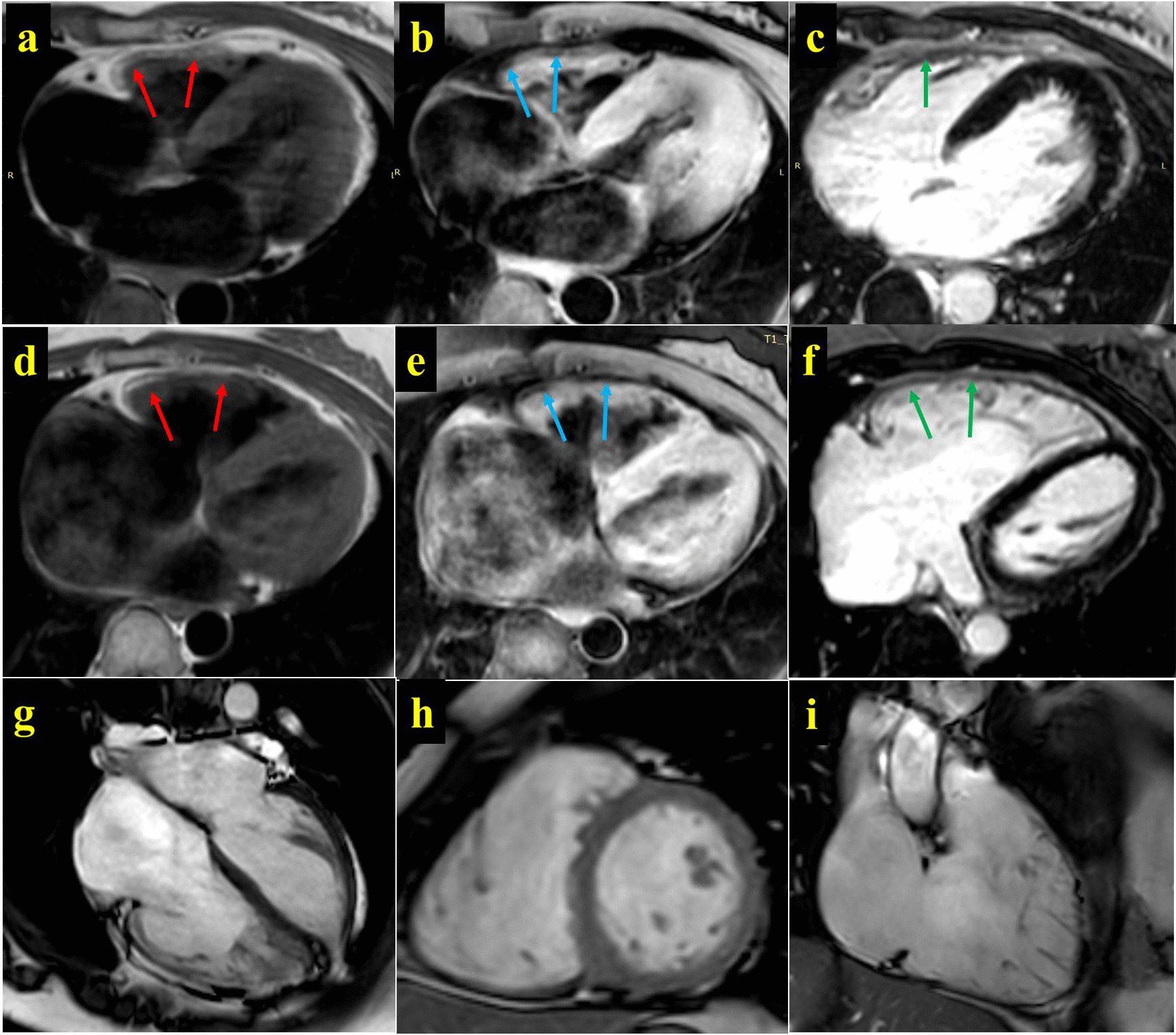
We discovered a Chinese Han family of ARVC/D with right ventricular HF (RVHF), cerebral thromboembolism, arrhythmias (atrial fibrillation, atrial standstill, multifocal ventricular premature, complete right bundle block and third-degree atrioventricular block) and sudden death. Based on the WES data, the variants of LMNA p.A242V, LAMA4 p.A225P and RYR2 p.T858M are highly conserved and predicated as "deleterious" by SIFT and MetaSVM algorithms. Their CADD predicting scores are 33, 27.4 and 25.8, respectively. These variants increase the hydrophobicity of their corresponding amino acid residues and their nearby sequences by 0.378, 0.266 and 0.289, respectively. The LAMA4 and RYR2 variants lead to changes in protein phosphorylation at or near their corresponding amino acid sites. There were high risks of joint pathogenicity for cardiomyopathy among these three variants. Cosegregation analysis indicated that LMNA p.A242V might be an important risk factor for ARVC/D, electrocardiogram abnormality and cerebral thromboembolism, while LAMA4 p.A225P may be a pathogenic etiology of ARVC/D and hereditary electrocardiogram abnormality.
The LMNA p.A242V may participate in the pathogenesis of familial ARVC/D with RVHF and cerebral thromboembolism, while LAMA4 p.A225P may be associated with ARVC/D and hereditary electrocardiogram abnormality.
致心律失常性右室心肌病/发育不良(ARVC/D)与室性心律失常、心力衰竭(HF)和心源性猝死有关。血栓栓塞也是 ARVC/D 的一个重要且严重的并发症。然而,ARVC/D 的病因及其与基因突变的关系尚不清楚。
对 ARVC/D 家系的外周血基因组 DNA 进行全外显子组测序(WES)和 Sanger 测序。然后,我们对易患心肌病和心律失常的基因进行了生物信息学分析。此外,我们还通过 ProtScale、NetPhos 和 ORVAL 算法分析了潜在的致病性突变如何影响氨基酸的疏水性和磷酸化以及它们的共同致病性。
我们发现了一个汉族 ARVC/D 家系,该家系表现为右心室 HF(RVHF)、脑栓塞、心律失常(心房颤动、心房停搏、多灶性室性早搏、完全性右束支阻滞和三度房室传导阻滞)和心源性猝死。基于 WES 数据,LMNA p.A242V、LAMA4 p.A225P 和 RYR2 p.T858M 的变异高度保守,且 SIFT 和 MetaSVM 算法预测为“有害”。它们的 CADD 预测评分分别为 33、27.4 和 25.8。这些变异分别使相应氨基酸残基及其附近序列的疏水性增加了 0.378、0.266 和 0.289。LAMA4 和 RYR2 变异导致相应氨基酸位点或其附近的蛋白质磷酸化发生变化。这三种变异的联合致病性对心肌病有较高的风险。共分离分析表明,LMNA p.A242V 可能是 ARVC/D、心电图异常和脑栓塞的重要危险因素,而 LAMA4 p.A225P 可能是 ARVC/D 和遗传性心电图异常的致病病因。
LMNA p.A242V 可能参与家族性 ARVC/D 伴 RVHF 和脑栓塞的发病机制,而 LAMA4 p.A225P 可能与 ARVC/D 和遗传性心电图异常有关。