Tulane Center for Biomedical Informatics and Genomics, Division of Biomedical Informatics and Genomics, John W. Deming Department of Medicine, School of Medicine, Tulane University, New Orleans, LA 70112, USA.
Department of Bioscience and Bioinformatics, Kyushu Institute of Technology, 680-4 Kawazu, Iizuka, Fukuoka 820-8502, Japan.
Mol Ther. 2022 Aug 3;30(8):2856-2867. doi: 10.1016/j.ymthe.2022.05.001. Epub 2022 May 6.
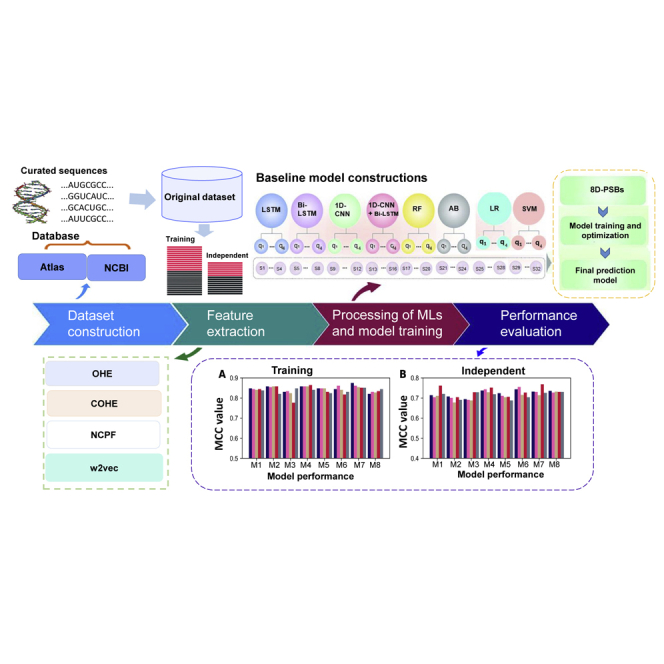
As one of the most prevalent post-transcriptional epigenetic modifications, N5-methylcytosine (m5C) plays an essential role in various cellular processes and disease pathogenesis. Therefore, it is important accurately identify m5C modifications in order to gain a deeper understanding of cellular processes and other possible functional mechanisms. Although a few computational methods have been proposed, their respective models have been developed using small training datasets. Hence, their practical application is quite limited in genome-wide detection. To overcome the existing limitations, we propose Deepm5C, a bioinformatics method for identifying RNA m5C sites throughout the human genome. To develop Deepm5C, we constructed a novel benchmarking dataset and investigated a mixture of three conventional feature-encoding algorithms and a feature derived from word-embedding approaches. Afterward, four variants of deep-learning classifiers and four commonly used conventional classifiers were employed and trained with the four encodings, ultimately obtaining 32 baseline models. A stacking strategy is effectively utilized by integrating the predicted output of the optimal baseline models and trained with a one-dimensional (1D) convolutional neural network. As a result, the Deepm5C predictor achieved excellent performance during cross-validation with a Matthews correlation coefficient and an accuracy of 0.697 and 0.855, respectively. The corresponding metrics during the independent test were 0.691 and 0.852, respectively. Overall, Deepm5C achieved a more accurate and stable performance than the baseline models and significantly outperformed the existing predictors, demonstrating the effectiveness of our proposed hybrid framework. Furthermore, Deepm5C is expected to assist community-wide efforts in identifying putative m5Cs and to formulate the novel testable biological hypothesis.
作为最普遍的转录后表观遗传修饰之一,N5-甲基胞嘧啶(m5C)在各种细胞过程和疾病发病机制中起着至关重要的作用。因此,准确识别 m5C 修饰对于深入了解细胞过程和其他可能的功能机制非常重要。尽管已经提出了几种计算方法,但它们各自的模型都是使用小型训练数据集开发的。因此,它们在全基因组检测中的实际应用非常有限。为了克服现有的局限性,我们提出了 Deepm5C,这是一种用于识别人类基因组中 RNA m5C 位点的生物信息学方法。为了开发 Deepm5C,我们构建了一个新的基准数据集,并研究了三种传统特征编码算法和一种来自词嵌入方法的特征的混合。之后,使用四种深度学习分类器和四种常用的传统分类器对这四种编码进行了训练,最终获得了 32 个基线模型。通过整合最优基线模型的预测输出并使用一维(1D)卷积神经网络进行训练,有效地利用了堆叠策略。结果,Deepm5C 在交叉验证中表现出色,马修斯相关系数和准确率分别为 0.697 和 0.855。在独立测试中的相应指标分别为 0.691 和 0.852。总的来说,Deepm5C 比基线模型实现了更准确和稳定的性能,并且显著优于现有的预测器,证明了我们提出的混合框架的有效性。此外,Deepm5C 有望协助全社区努力识别推定的 m5C 并提出新的可测试生物学假设。