School of Life Sciences, University of Nottingham, Nottingham, United Kingdom.
DeepSeq, Centre for Genetics and Genomics, Queen's Medical Centre, University of Nottingham, Nottingham, United Kingdom.
Front Cell Infect Microbiol. 2022 Apr 5;12:841138. doi: 10.3389/fcimb.2022.841138. eCollection 2022.
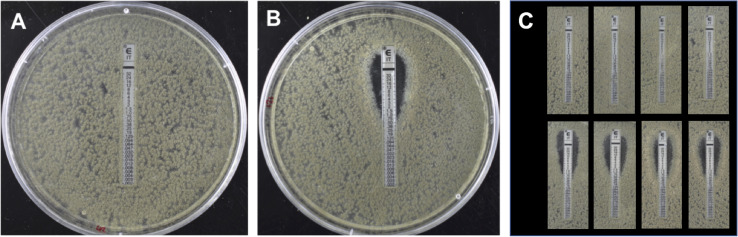
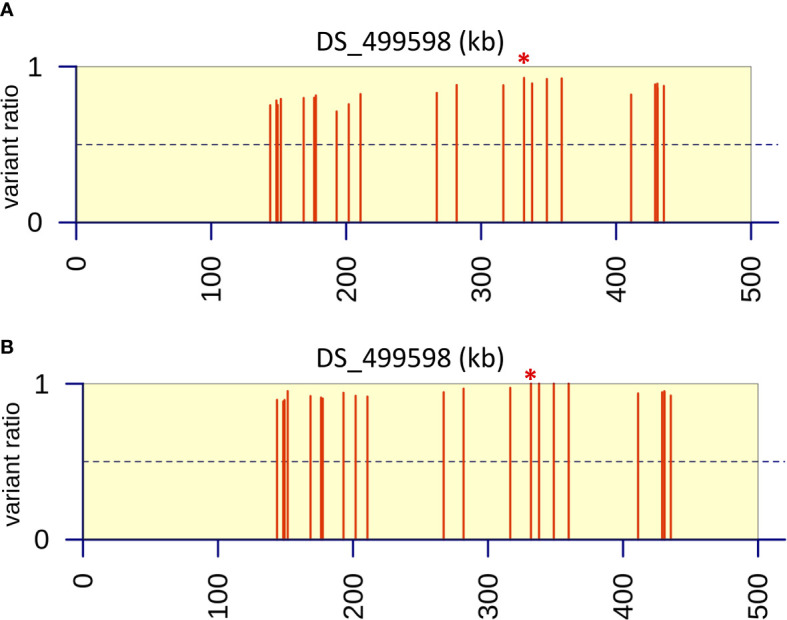
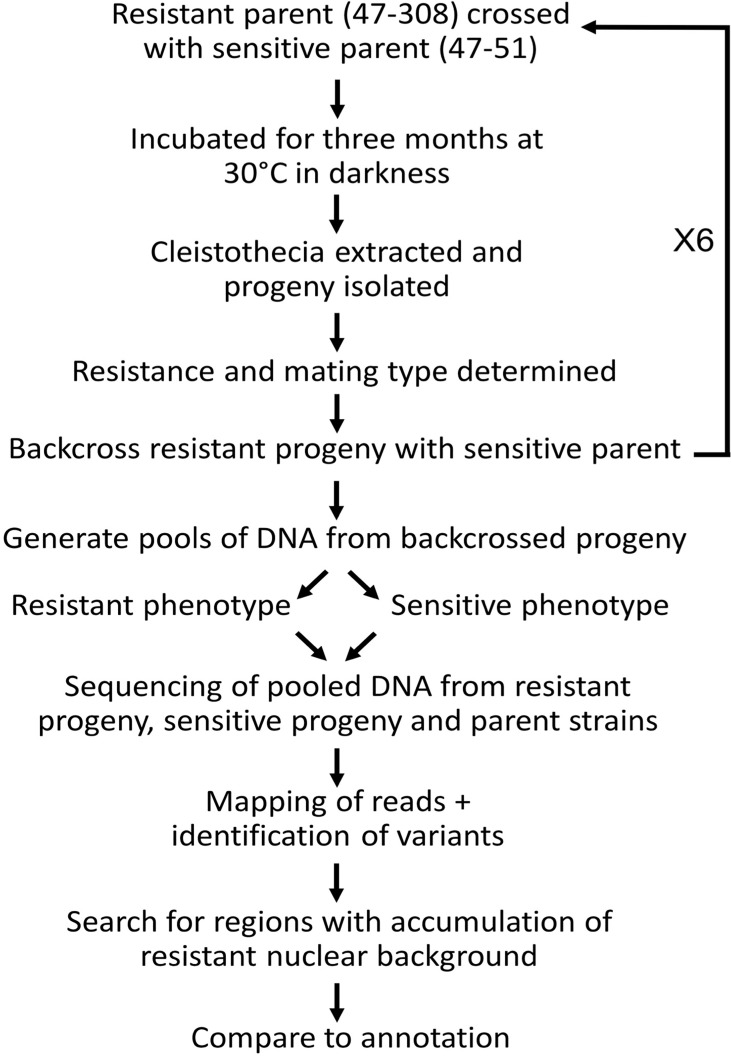
A sexual cycle was described in 2009 for the opportunistic fungal pathogen , opening up for the first time the possibility of using techniques reliant on sexual crossing for genetic analysis. The present study was undertaken to evaluate whether the technique 'bulk segregant analysis' (BSA), which involves detection of differences between pools of progeny varying in a particular trait, could be applied in conjunction with next-generation sequencing to investigate the underlying basis of monogenic traits in . Resistance to the azole antifungal itraconazole was chosen as a model, with a dedicated bioinformatic pipeline developed to allow identification of SNPs that differed between the resistant progeny pool and resistant parent compared to the sensitive progeny pool and parent. A clinical isolate exhibiting monogenic resistance to itraconazole of unknown basis was crossed to a sensitive parent and F1 progeny used in BSA. In addition, the use of backcrossing and increasing the number in progeny pools was evaluated as ways to enhance the efficiency of BSA. Use of F1 pools of 40 progeny led to the identification of 123 candidate genes with SNPs distributed over several contigs when aligned to an A1163 reference genome. Successive rounds of backcrossing enhanced the ability to identify specific genes and a genomic region, with BSA of progeny (using 40 per pool) from a third backcross identifying 46 genes with SNPs, and BSA of progeny from a sixth backcross identifying 20 genes with SNPs in a single 292 kb region of the genome. The use of an increased number of 80 progeny per pool also increased the resolution of BSA, with 29 genes demonstrating SNPs between the different sensitive and resistant groupings detected using progeny from just the second backcross with the majority of variants located on the same 292 kb region. Further bioinformatic analysis of the 292 kb region identified the presence of a gene variant resulting in a methionine to lysine (M220K) change in the CYP51A protein, which was concluded to be the causal basis of the observed resistance to itraconazole. The future use of BSA in genetic analysis of is discussed.
2009 年,人们描述了一种性循环,为首次利用依赖于有性杂交的技术进行遗传分析开辟了可能性。本研究旨在评估“大量分离分析”(BSA)技术是否可以与下一代测序相结合应用于研究单基因性状的基础。选择唑类抗真菌药物伊曲康唑的抗性作为模型,开发了专门的生物信息学管道,以鉴定在抗性后代池与抗性亲本之间与敏感后代池和亲本之间存在差异的 SNPs。使用表现出未知唑类抗药性的单基因抗性临床分离株与敏感亲本杂交,并在 BSA 中使用 F1 后代。此外,还评估了回交和增加后代池数量作为提高 BSA 效率的方法。使用 40 个后代的 F1 池导致在与 A1163 参考基因组比对时,分布在几个连续体上的 123 个候选基因的鉴定,这些基因具有 SNPs。连续回交增强了鉴定特定基因和基因组区域的能力,来自第三次回交的 40 个后代的 BSA 鉴定了 46 个具有 SNPs 的基因,来自第六次回交的 40 个后代的 BSA 鉴定了 20 个具有 SNPs 的基因在基因组的单个 292kb 区域中。使用每个池增加到 80 个后代也增加了 BSA 的分辨率,使用仅来自第二次回交的后代检测到不同敏感和抗性分组之间的 29 个基因具有 SNPs,大多数变体位于相同的 292kb 区域。对 292kb 区域的进一步生物信息学分析确定了存在一个基因变体,导致 CYP51A 蛋白中的蛋氨酸到赖氨酸(M220K)变化,该变体被认为是观察到的伊曲康唑抗性的因果基础。讨论了 BSA 在 遗传分析中的未来应用。