Department of Lysosomal Diseases, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico.
Department of Pediatrics, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico.
Mol Genet Genomic Med. 2022 Jul;10(7):e1957. doi: 10.1002/mgg3.1957. Epub 2022 May 9.
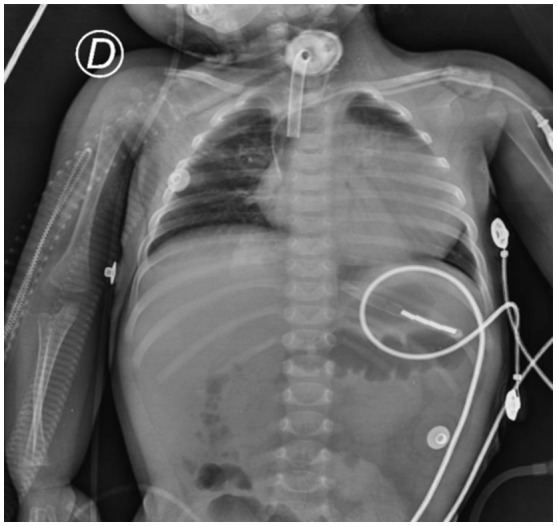
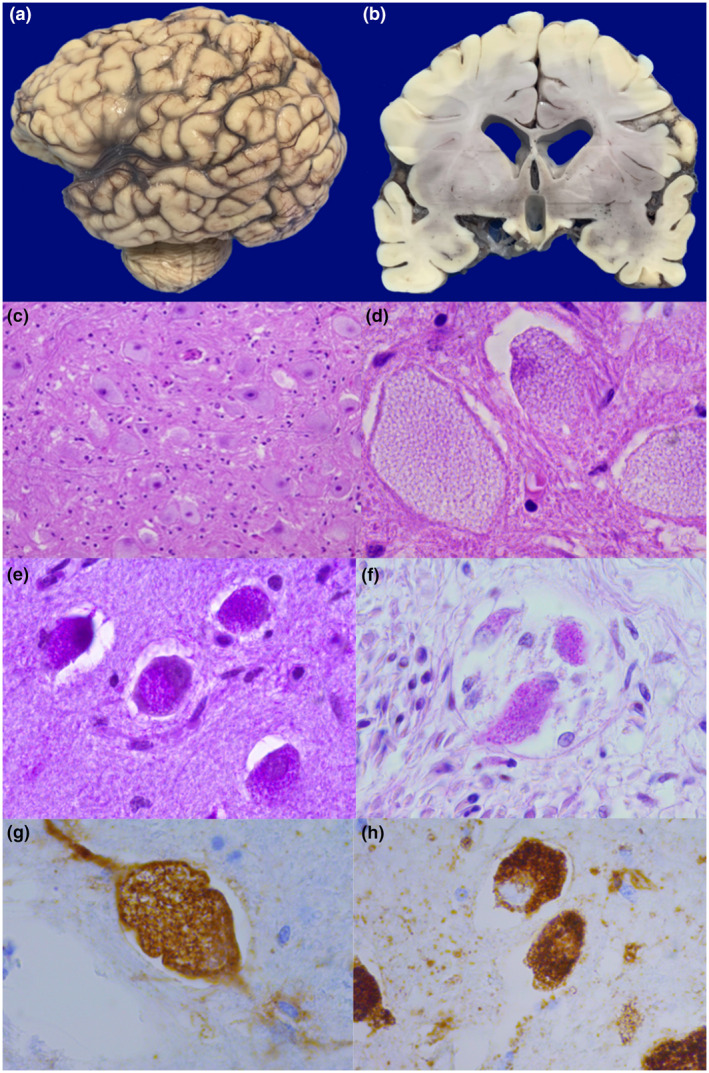
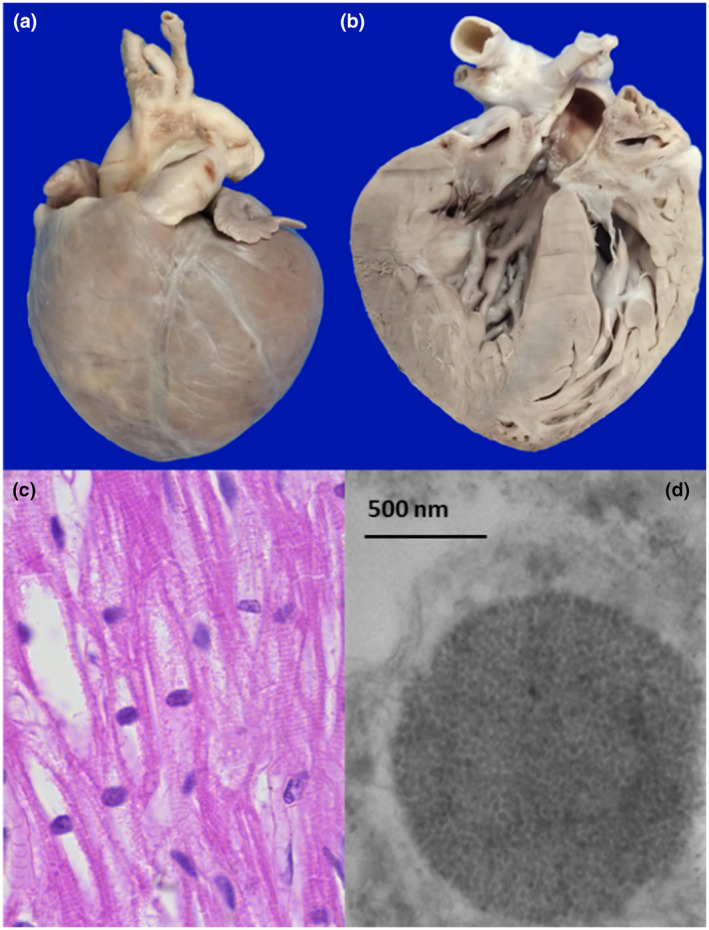
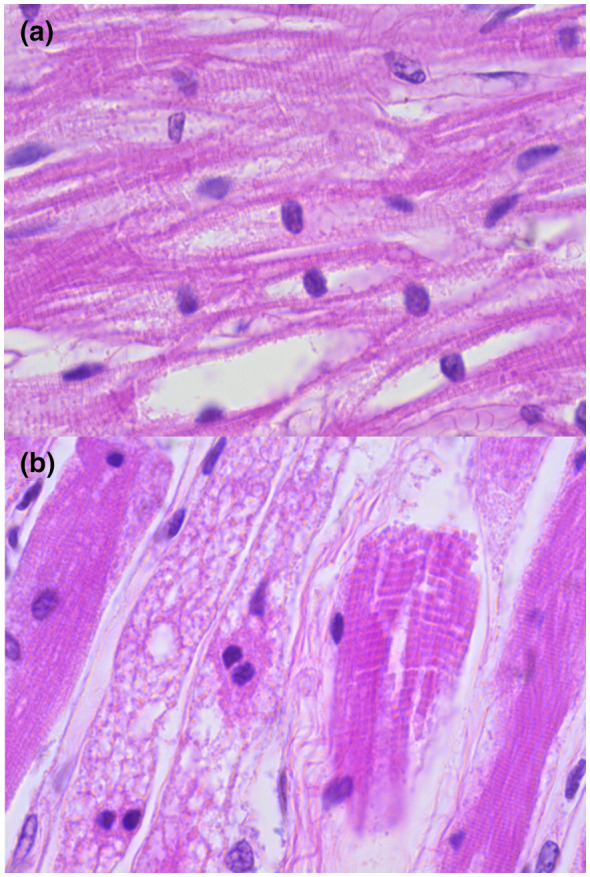
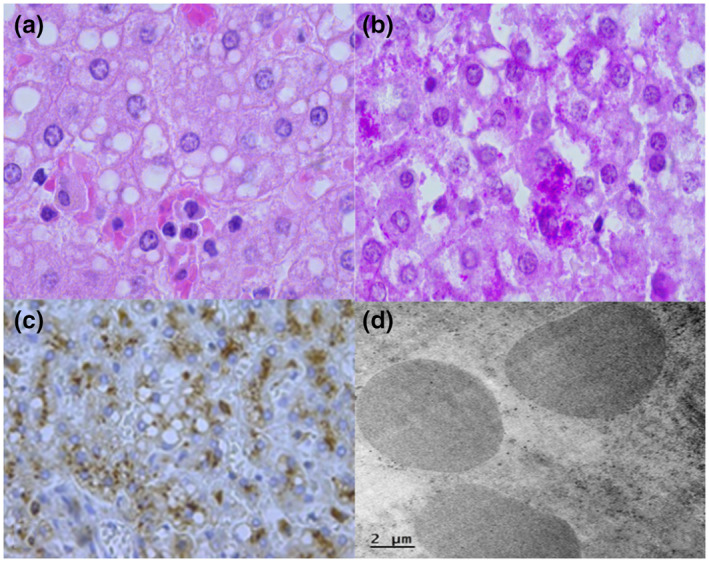
Pompe disease (PD) is an autosomal recessive disorder by a deficiency of acid α-glucosidase (GAA) with intralysosomal glycogen accumulation in multiple tissues. We present the case of a 5-month-old male with hypertrophic cardiomyopathy, hypotony, feeding difficulties, and oxygen requirement since birth. At 3 months of age, he develops heart failure, respiratory impairment, and neurological deterioration. The echocardiogram revealed concentric hypertrophic cardiomyopathy with left-diastolic dysfunction. We found increased creatine-phosphokinase, lactate dehydrogenase, and urinary glucose tetrasaccharide levels, 50% of PAS-positive vacuolated lymphocytes in the peripheral blood smear, and low GAA activity. Sequencing of coding exons and flanking intronic sequences revealed a novel homozygous 4 bp deletion in exon 15 of the GAA gene (c.2066_2069delAGCC/p.Glu689Glyfs*6). IOPD was diagnosed. At 5 months old, we started enzyme replacement therapy with an alpha-alglucosidase of 20 mg/kg weekly and immunomodulation with intravenous immunoglobulin. He developed two cardiorespiratory arrests with subsequent neurologic deterioration, convulsive crisis, and respiratory failure and died at 9 months old. We found the usual PD hallmarks in the heart, striated muscle, and liver but also we found neuronal lesions characterized by cytoplasm vacuolization with PAS-positive granules in the central nervous system and myenteric plexus. We describe a novel GAA gene pathogenic variant with a particular phenotype characterized by classic IOPD and neurologic histopathological findings. Enhancing the knowledge of lysosomal diseases is critical to improving the diagnosis and treatment of these patients.
庞贝病(PD)是一种常染色体隐性遗传病,由于酸性α-葡萄糖苷酶(GAA)缺乏,导致溶酶体内糖原堆积,累及多种组织。我们报告了一例 5 月龄男性患儿,起病表现为肥厚型心肌病、眼压低、喂养困难和出生后即需吸氧。3 月龄时出现心力衰竭、呼吸功能障碍和神经功能恶化。超声心动图显示为向心性肥厚型心肌病伴左心室舒张功能障碍。我们发现患儿肌酸磷酸激酶、乳酸脱氢酶和尿葡萄糖四糖水平升高,外周血涂片可见 50% PAS 阳性空泡淋巴细胞,GAA 活性降低。编码外显子和侧翼内含子序列的测序显示 GAA 基因第 15 外显子中存在一个新的纯合性 4bp 缺失(c.2066_2069delAGCC/p.Glu689Glyfs*6)。诊断为婴儿型庞贝病。5 月龄时开始每周给予 20mg/kg 的 α-葡萄糖苷酶进行酶替代治疗,并联合静脉用免疫球蛋白进行免疫调节。患儿发生 2 次心肺骤停,随后出现神经功能恶化、惊厥发作和呼吸衰竭,9 月龄时死亡。我们在心脏、横纹肌和肝脏中发现了 PD 的典型组织学特征,还在中枢神经系统和肌间神经丛中发现了神经元病变,其特征为细胞质空泡化,PAS 阳性颗粒。我们描述了一种新的 GAA 基因突变,其表型独特,具有典型的 IOPD 和神经组织病理学表现。提高对溶酶体疾病的认识对于改善这些患者的诊断和治疗至关重要。