Manchester Institute of Biotechnology, The University of Manchester, 131 Princess Street, Manchester M1 7DN, United Kingdom.
Department of Chemical Engineering, The University of Manchester, Oxford Road, Manchester M13 9PL, United Kingdom.
J Am Chem Soc. 2022 Jun 22;144(24):10752-10767. doi: 10.1021/jacs.2c01375. Epub 2022 May 10.
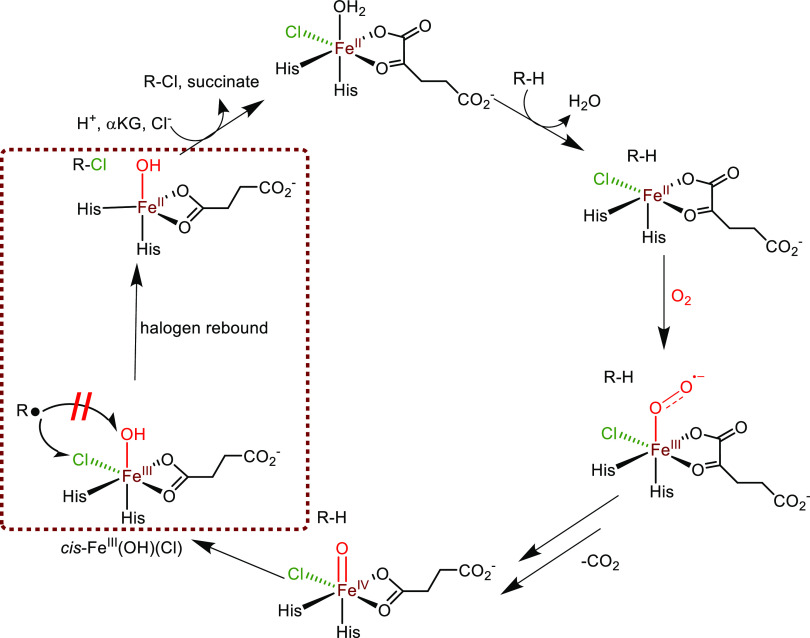
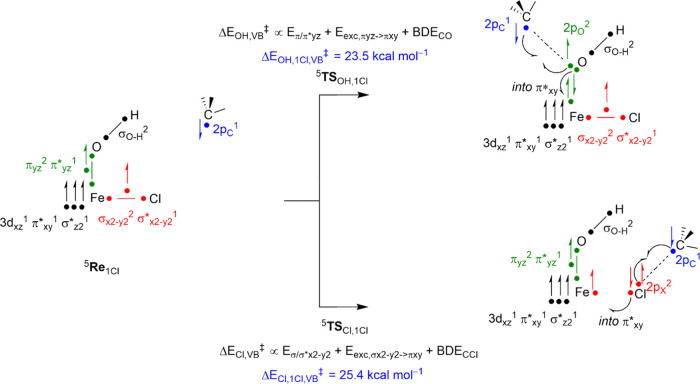
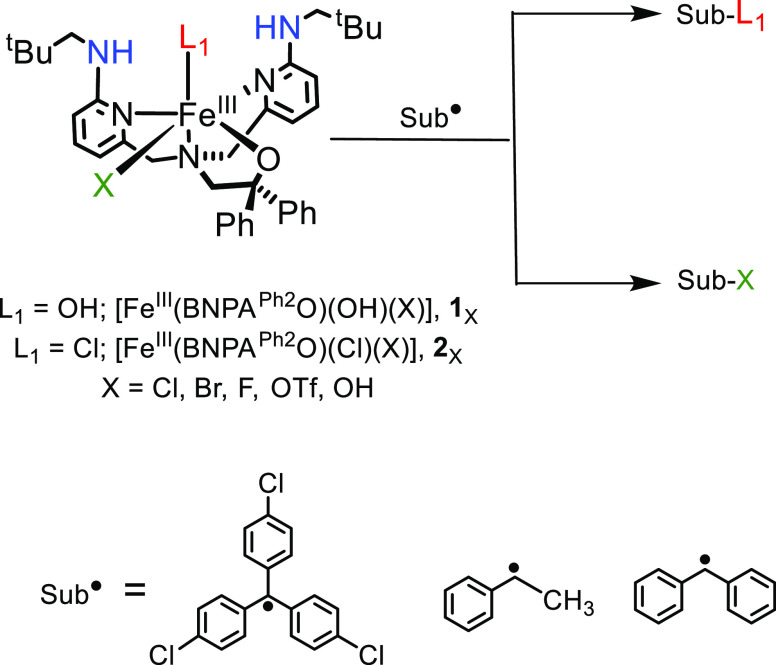
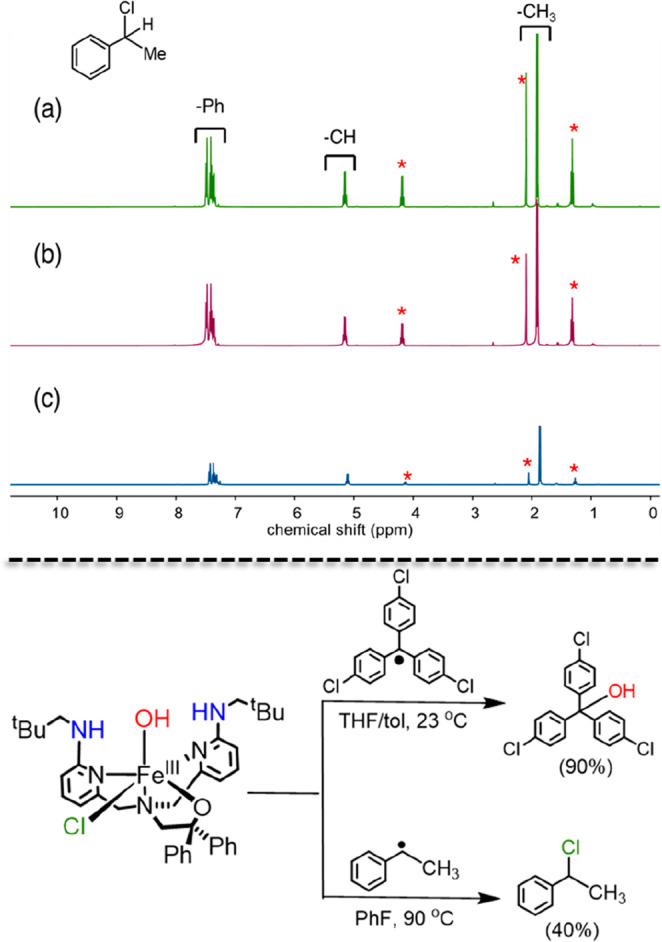
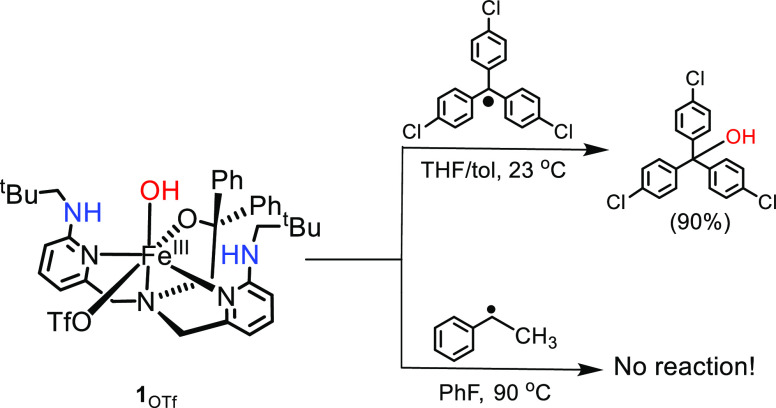
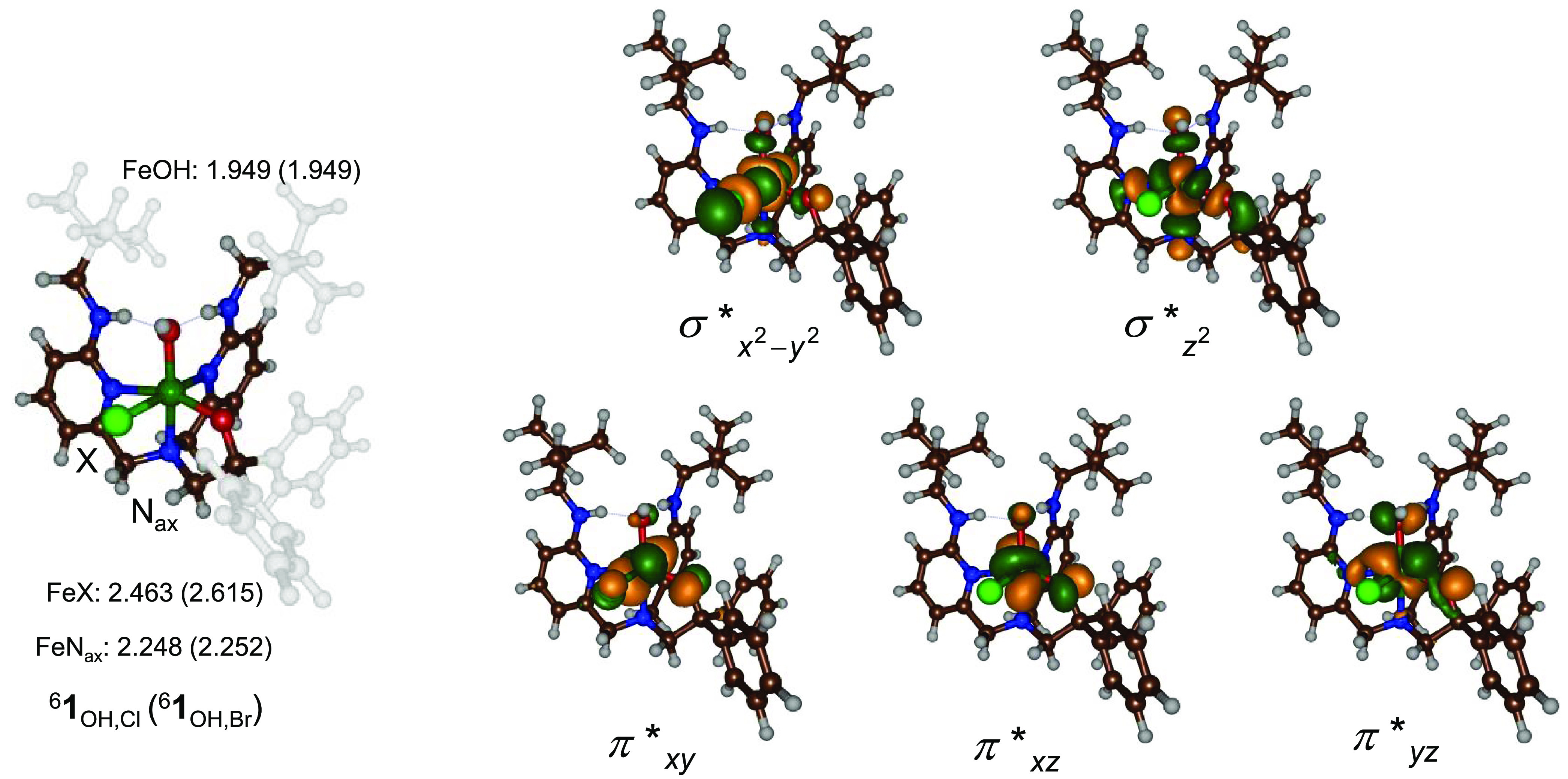
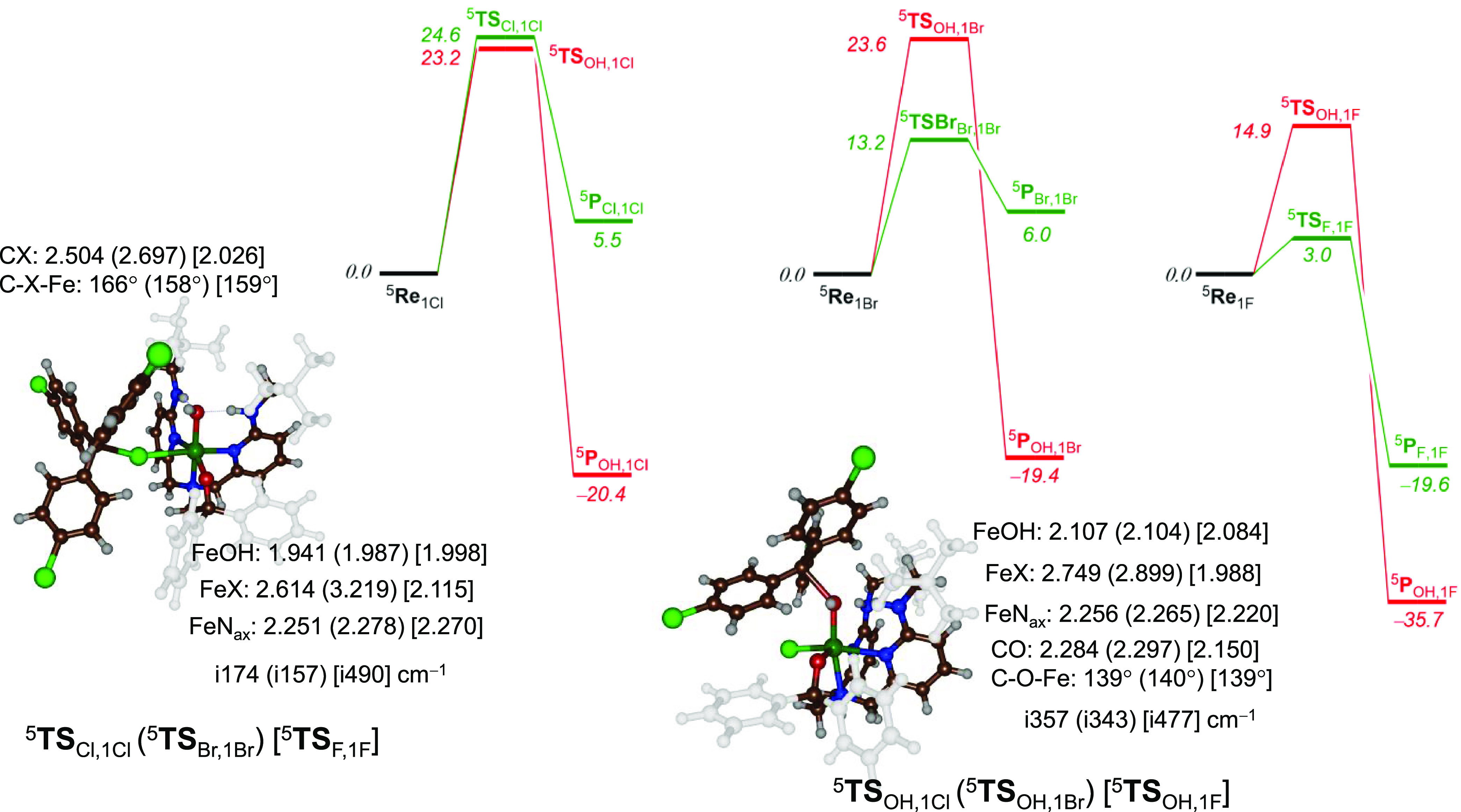
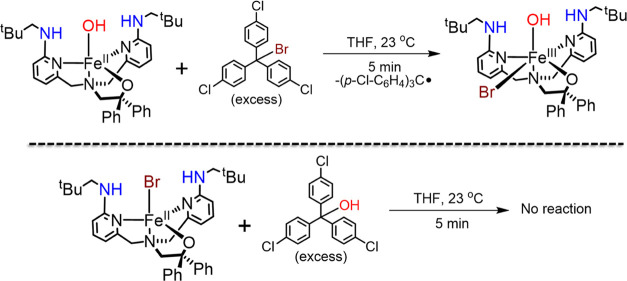
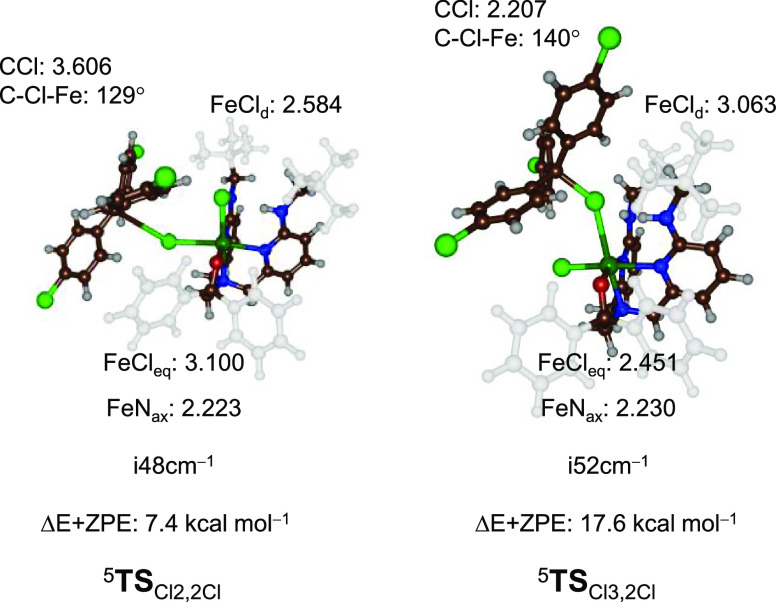
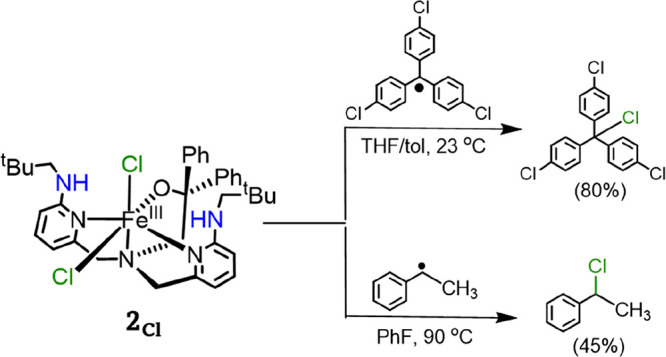
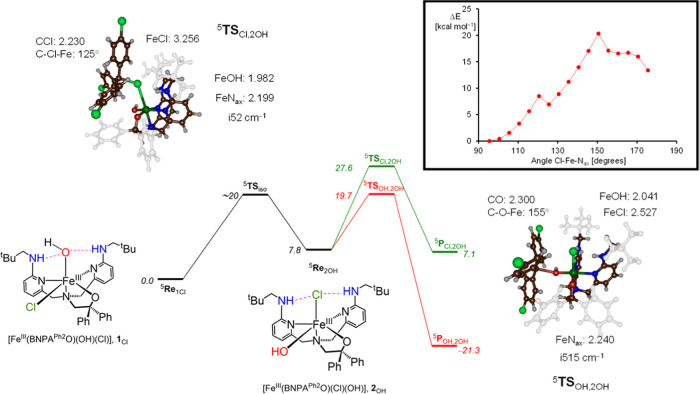
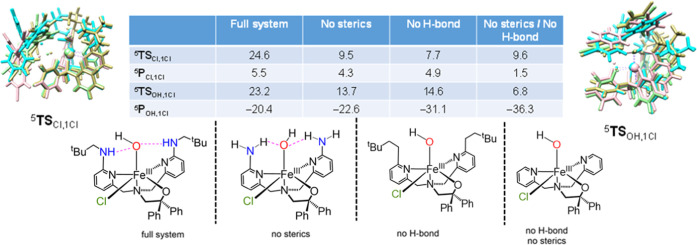
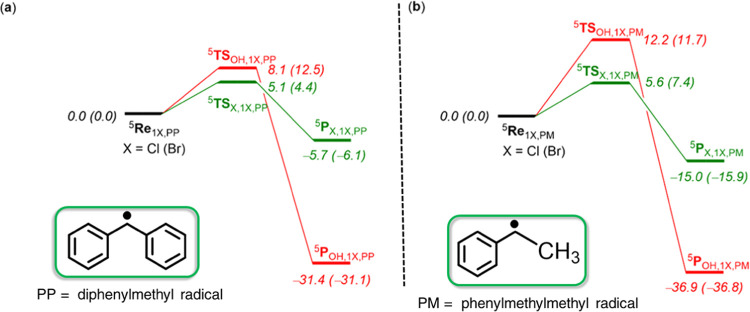
Nonheme iron halogenases are unique enzymes in nature that selectively activate an aliphatic C-H bond of a substrate to convert it into C-X (X = Cl/Br, but not F/I). It is proposed that they generate an Fe(OH)(X) intermediate in their catalytic cycle. The analogous Fe(OH) intermediate in nonheme iron hydroxylases transfers OH to give alcohol product, whereas the halogenases transfer X to the carbon radical substrate. There remains significant debate regarding what factors control their remarkable selectivity of the halogenases. The reactivity of the complexes Fe(BNPAO)(OH)(X) (X = Cl, Br) with a secondary carbon radical (R) is described. It is found that X transfer occurs with a secondary carbon radical, as opposed to OH transfer with tertiary radicals. Comprehensive computational studies involving density functional theory were carried out to examine the possible origins of this selectivity. The calculations reproduce the experimental findings, which indicate that halogen transfer is not observed for the tertiary radicals because of a nonproductive equilibrium that results from the endergonic nature of these reactions, despite a potentially lower reaction barrier for the halogenation pathway. In contrast, halogen transfer is favored for secondary carbon radicals, for which the halogenated product complex is thermodynamically more stable than the reactant complex. These results are rationalized by considering the relative strengths of the C-X bonds that are formed for tertiary versus secondary carbon centers. The computational analysis also shows that the reaction barrier for halogen transfer is significantly dependent on secondary coordination sphere effects, including steric and H-bonding interactions.
非血红素铁卤代酶是自然界中独特的酶,能够选择性地激活底物的脂肪族 C-H 键,将其转化为 C-X(X=Cl/Br,但不是 F/I)。据推测,它们在催化循环中生成 Fe(OH)(X)中间体。非血红素铁羟化酶中的类似 Fe(OH)中间体将 OH 转移到产物中形成醇,而卤代酶则将 X 转移到碳自由基底物上。对于控制卤代酶显著选择性的因素,仍存在很大争议。描述了配合物 Fe(BNPAO)(OH)(X)(X=Cl,Br)与仲碳自由基(R)的反应性。发现 X 转移发生在仲碳自由基上,而不是叔碳自由基上的 OH 转移。进行了涉及密度泛函理论的综合计算研究,以研究这种选择性的可能起源。计算结果再现了实验结果,表明对于叔碳自由基,不会观察到卤素转移,这是由于这些反应的内禀能性质导致非生产性平衡,尽管卤化途径的反应势垒可能较低。相比之下,卤素转移有利于仲碳自由基,因为卤代产物配合物比反应物配合物热力学上更稳定。通过考虑形成叔碳和仲碳中心的 C-X 键的相对强度,可以合理地解释这些结果。计算分析还表明,卤素转移的反应势垒高度依赖于次级配位球效应,包括空间位阻和氢键相互作用。