Wasalathilake Kimal Chandula, Roknuzzaman Md, Ostrikov Kostya Ken, Ayoko Godwin A, Yan Cheng
School of Chemistry, Physics and Mechanical Engineering, Faculty of Science and Engineering, Queensland University of Technology (QUT) Brisbane QLD 4001 Australia
RSC Adv. 2018 Jan 9;8(5):2271-2279. doi: 10.1039/c7ra11628d.
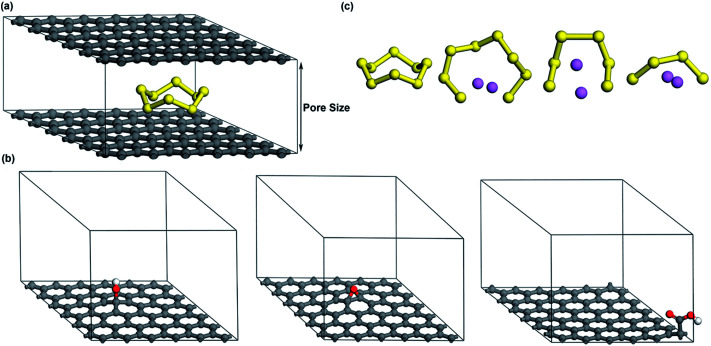
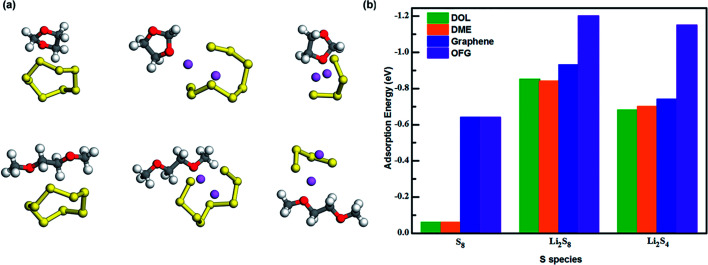
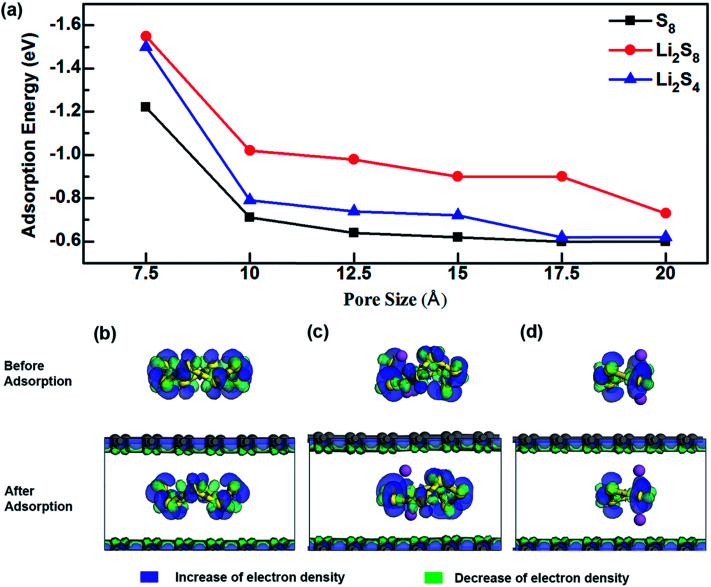
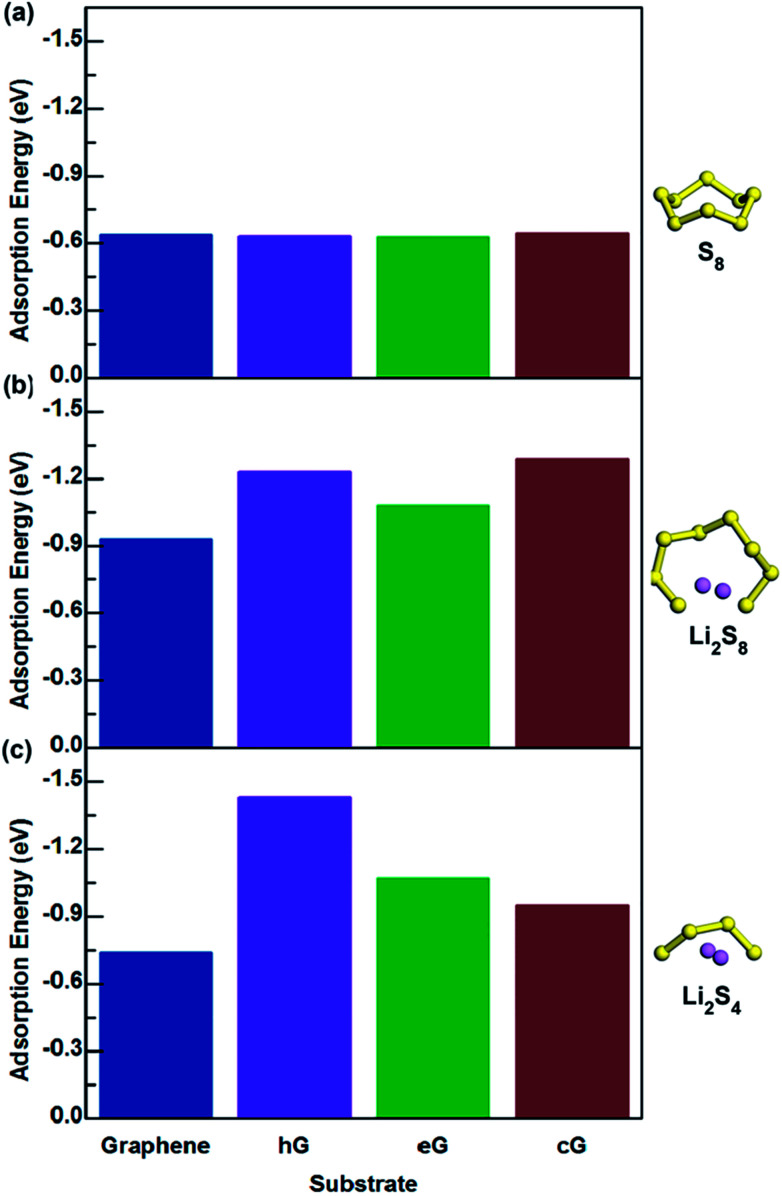
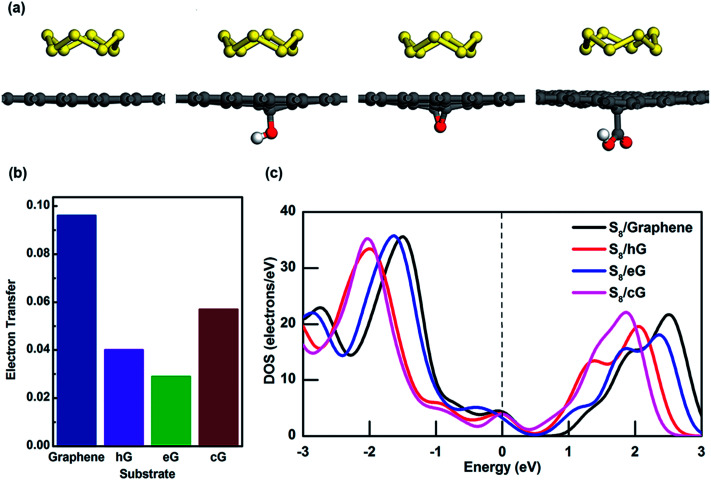
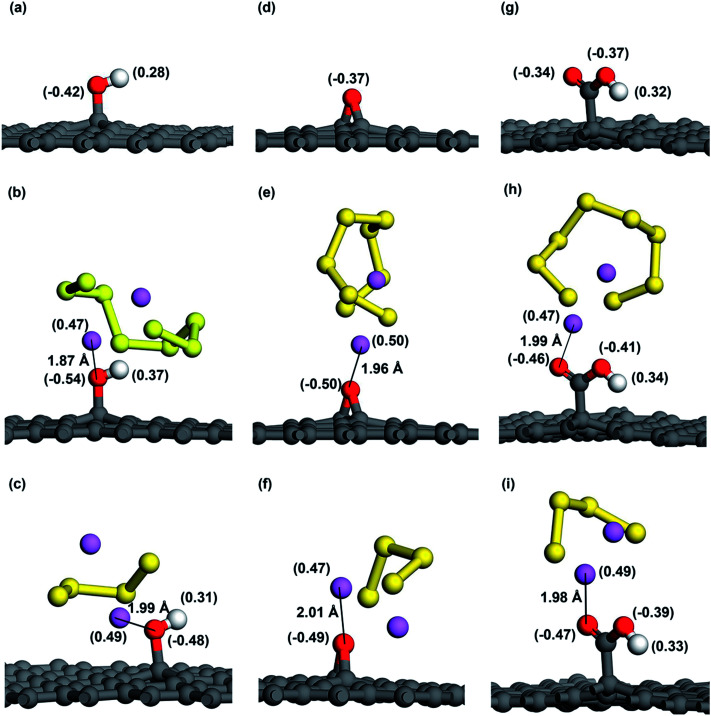
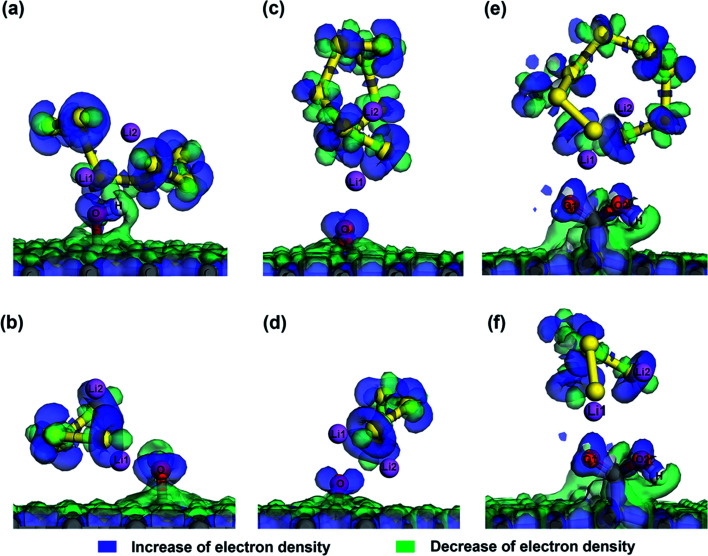
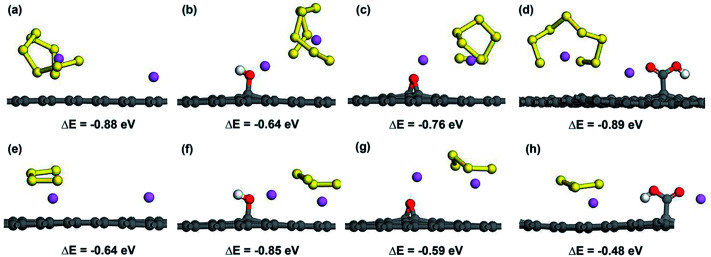
Lithium-sulfur (Li-S) batteries are emerging as one of the promising candidates for next generation rechargeable batteries. However, dissolution of lithium polysulfides in the liquid electrolyte, low electrical conductivity of sulfur and large volume change during electrochemical cycling are the main technical challenges for practical applications. In this study, a systematic first-principles density functional theory calculation is adopted to understand the interactions between graphene and graphene with oxygen containing functional groups (hydroxyl, epoxy and carboxyl groups) and sulphur (S) and long chain lithium polysulfides (LiS and LiS). We find the adsorption is dominated by different mechanisms in sulphur and lithium polysulfides, van der Waals attraction and formation of coordinate covalent Li-O bonds. The adsorption strength is dependent on the inter-layer distance and electron rich functional groups. Through these mechanisms, sulphur and lithium polysulfides can be successfully retained in porous graphene, leading to improved conductivity and charge transfer in the cathode of Li-S batteries.
锂硫(Li-S)电池正成为下一代可充电电池中颇具前景的候选者之一。然而,多硫化锂在液体电解质中的溶解、硫的低电导率以及电化学循环过程中的大体积变化是实际应用中的主要技术挑战。在本研究中,采用了系统的第一性原理密度泛函理论计算,以了解石墨烯与含氧基官能团(羟基、环氧基和羧基)的石墨烯与硫(S)以及长链多硫化锂(LiS和LiS)之间的相互作用。我们发现,在硫和多硫化锂中,吸附由不同的机制主导,即范德华引力和配位共价Li-O键的形成。吸附强度取决于层间距离和富电子官能团。通过这些机制,硫和多硫化锂可以成功地保留在多孔石墨烯中,从而提高Li-S电池阴极的电导率和电荷转移。