Institute of Human Genetics, University Medical Center Göttingen, Göttingen, Germany.
Institute for Auditory Neuroscience and InnerEarLab, University Medical Center Göttingen, Göttingen, Germany.
J Eur Acad Dermatol Venereol. 2022 Sep;36(9):1606-1611. doi: 10.1111/jdv.18207. Epub 2022 May 25.
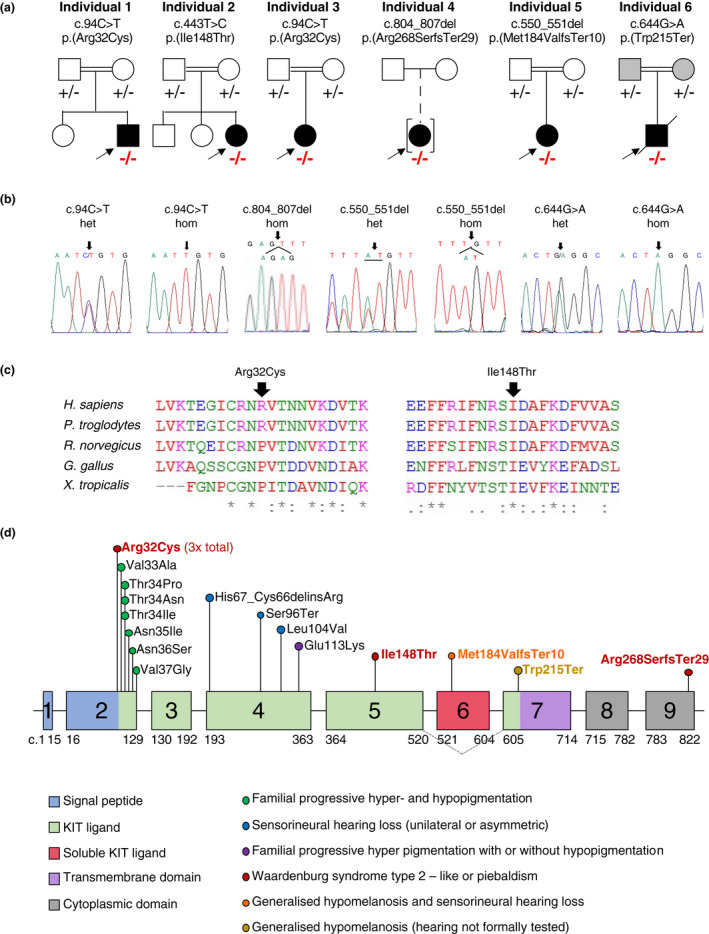
Pathogenic variants in KITLG, a crucial protein involved in pigmentation and neural crest cell migration, cause non-syndromic hearing loss, Waardenburg syndrome type 2, familial progressive hyperpigmentation and familial progressive hyper- and hypopigmentation, all of which are inherited in an autosomal dominant manner.
To describe the genotypic and clinical spectrum of biallelic KITLG-variants.
We used a genotype-first approach through the GeneMatcher data sharing platform to collect individuals with biallelic KITLG variants and reviewed the literature for overlapping reports.
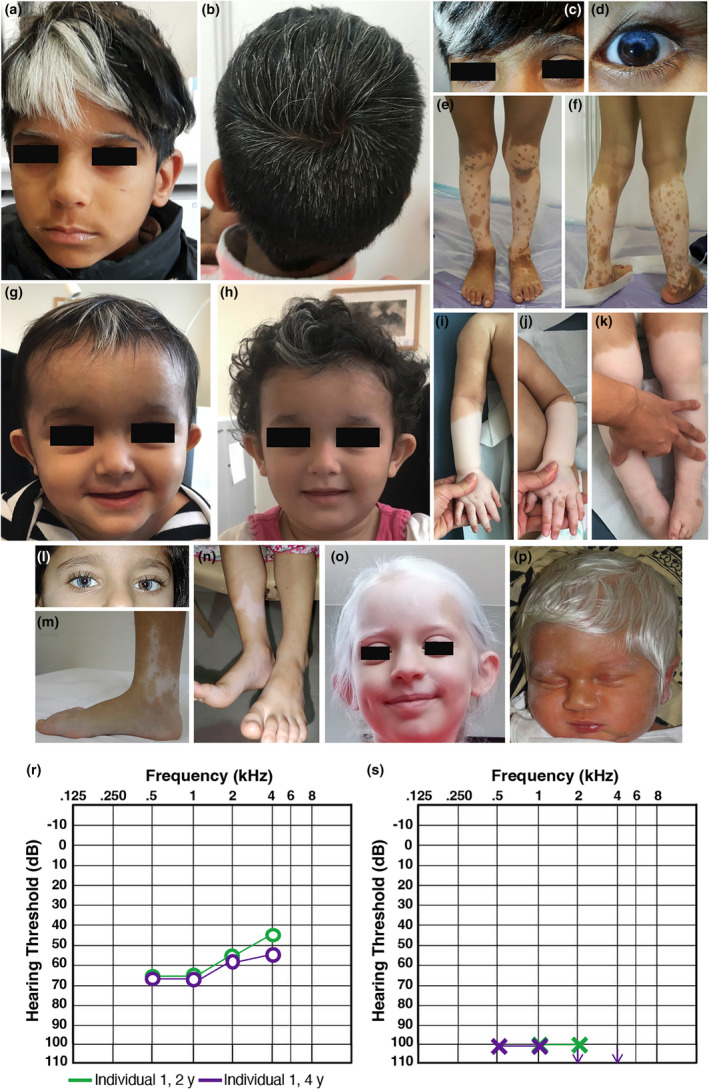
We describe the first case series with biallelic KITLG variants; we expand the known hypomelanosis spectrum to include a 'sock-and-glove-like', symmetric distribution, progressive repigmentation and generalized hypomelanosis. We speculate that KITLG biallelic loss-of-function variants cause generalized hypomelanosis, whilst variants with residual function lead to a variable auditory-pigmentary disorder mostly reminiscent of Waardenburg syndrome type 2 or piebaldism.
We provide consolidating evidence that biallelic KITLG variants cause a distinct auditory-pigmentary disorder. We evidence a significant clinical variability, similar to the one previously observed in KIT-related piebaldism.
KITLG 是一种关键的蛋白质,参与色素沉着和神经嵴细胞迁移,其致病性变异可导致非综合征性听力损失、Waardenburg 综合征 2 型、家族性进行性色素沉着过度和家族性进行性色素沉着减少/增多,所有这些疾病均以常染色体显性遗传方式遗传。
描述双等位基因 KITLG 变异的基因型和临床谱。
我们通过 GeneMatcher 数据共享平台采用先基因型后表型的方法,收集双等位基因 KITLG 变异的个体,并对重叠报告的文献进行回顾。
我们描述了首例双等位基因 KITLG 变异的病例系列;我们将已知的色素减退谱扩展至包括“袜子和手套样”、对称分布、进行性复色和泛发性色素减退。我们推测 KITLG 双等位基因失活变异导致泛发性色素减退,而具有残留功能的变异导致以 Waardenburg 综合征 2 型或斑驳病为主要表现的可变的听觉-色素障碍。
我们提供了确凿的证据表明,双等位基因 KITLG 变异可导致独特的听觉-色素障碍。我们发现了显著的临床变异性,与之前观察到的 KIT 相关斑驳病相似。