Xiao Tiantian, Chen Xiang, Xu Yan, Chen Huiyao, Dong Xinran, Yang Lin, Wu Bingbing, Chen Liping, Li Long, Zhuang Deyi, Chen Dongmei, Zhou Yuanfeng, Wang Huijun, Zhou Wenhao
Department of Neonatology, National Children's Medical Center, Children's Hospital of Fudan University, Shanghai, China.
Division of Neurology, National Children's Medical Center, Children's Hospital of Fudan University, Shanghai, China.
Front Mol Neurosci. 2022 Apr 26;15:809810. doi: 10.3389/fnmol.2022.809810. eCollection 2022.
-related disorder is typically characterized as neonatal onset seizure and epileptic encephalopathy. The relationship between its phenotype and genotype is still elusive. This study aims to provide clinical features, management, and prognosis of patients with novel candidate variants of the gene.
We enrolled patients with novel variants in the gene from the China Neonatal Genomes Project between January 2018 and January 2021. All patients underwent next-generation sequencing tests and genetic data were analyzed by an in-house pipeline. The pathogenicity of variants was classified according to the guideline of the American College of Medical Genetics. Each case was evaluated by two geneticists back to back. Patients' information was acquired from clinical records.
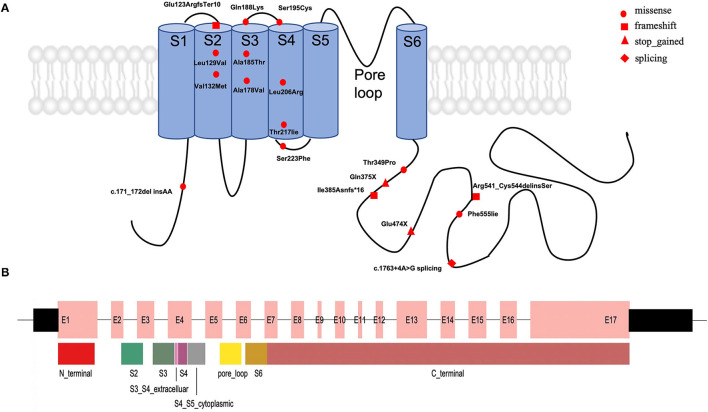
A total of 30 unrelated patients with novel variants in the gene were identified, including 19 patients with single-nucleotide variants (SNVs) and 11 patients with copy number variants (CNVs). For the 19 SNVs, 12 missense variants and 7 truncating variants were identified. Of them, 36.8% (7/19) of the variants were located in C-terminal regions, 15.7% (3/19) in segment S2, and 15.7% (3/19) in segment S4. Among them, 18 of 19 patients experienced seizures in the early neonatal period. However, one patient presented neurodevelopmental delay (NDD) as initial phenotype when he was 2 months old, and he had severe NDD when he was 3 years old. This patient did not present seizure but had abnormal electrographic background activity and brain imaging. Moreover, for the 11 patients with CNVs, 20q13.3 deletions involving , and genes were detected. All of them presented neonatal-onset seizures, responded to antiepileptic drugs, and had normal neurological development.
In this study, patients with novel variants have variable phenotypes, whereas patients with 20q13.3 deletion involving , and genes tend to have normal neurological development.
-相关疾病通常表现为新生儿期发作的癫痫和癫痫性脑病。其表型与基因型之间的关系仍不明确。本研究旨在提供具有该基因新型候选变异的患者的临床特征、治疗及预后情况。
我们纳入了2018年1月至2021年1月期间中国新生儿基因组计划中具有该基因新型变异的患者。所有患者均接受了下一代测序检测,基因数据通过内部流程进行分析。变异的致病性根据美国医学遗传学学会的指南进行分类。每个病例由两位遗传学家背对背评估。患者信息从临床记录中获取。
共鉴定出30例具有该基因新型变异的无关患者,其中19例为单核苷酸变异(SNV)患者和11例为拷贝数变异(CNV)患者。对于19个SNV,鉴定出12个错义变异和7个截短变异。其中,36.8%(7/19)的变异位于C端区域,15.7%(3/19)位于S2段,15.7%(3/19)位于S4段。其中,19例患者中有18例在新生儿早期出现癫痫发作。然而,1例患者在2个月大时以神经发育迟缓(NDD)为初始表型,3岁时出现严重NDD。该患者未出现癫痫发作,但脑电图背景活动和脑成像异常。此外,对于11例CNV患者,检测到涉及、和基因的20q13.3缺失。他们均表现为新生儿期发作的癫痫,对抗癫痫药物有反应,且神经发育正常。
在本研究中,具有该基因新型变异的患者具有可变的表型,而涉及、和基因的20q13.3缺失的患者往往神经发育正常。