Canada's Michael Smith Genome Sciences Center, Vancouver, BC, Canada.
Bioinformatics Graduate Program, University of British Columbia, Vancouver, BC, Canada.
Curr Protoc. 2022 May;2(5):e442. doi: 10.1002/cpz1.442.
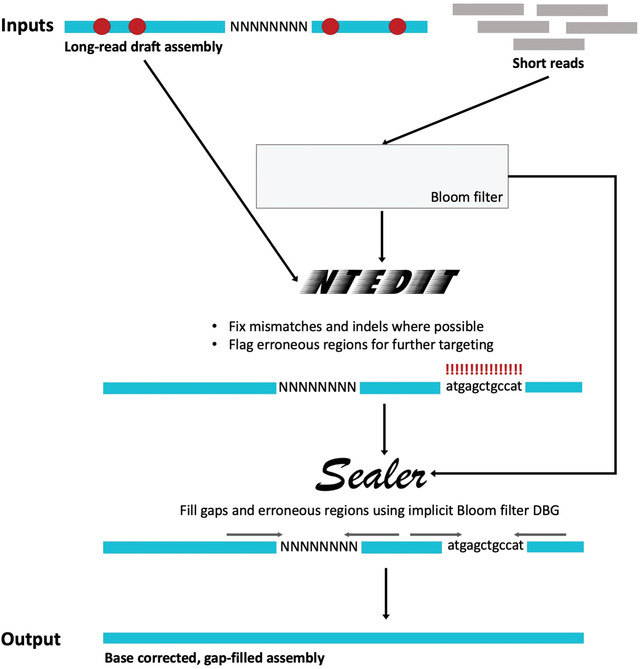
High-quality genome assemblies are crucial to many biological studies, and utilizing long sequencing reads can help achieve higher assembly contiguity. While long reads can resolve complex and repetitive regions of a genome, their relatively high associated error rates are still a major limitation. Long reads generally produce draft genome assemblies with lower base quality, which must be corrected with a genome polishing step. Hybrid genome polishing solutions can greatly improve the quality of long-read genome assemblies by utilizing more accurate short reads to validate bases and correct errors. Currently available hybrid polishing methods rely on read alignments, and are therefore memory-intensive and do not scale well to large genomes. Here we describe ntEdit+Sealer, an alignment-free, k-mer-based genome finishing protocol that employs memory-efficient Bloom filters. The protocol includes ntEdit for correcting base errors and small indels, and for marking potentially problematic regions, then Sealer for filling both assembly gaps and problematic regions flagged by ntEdit. ntEdit+Sealer produces highly accurate, error-corrected genome assemblies, and is available as a Makefile pipeline from https://github.com/bcgsc/ntedit_sealer_protocol. © 2022 The Authors. Current Protocols published by Wiley Periodicals LLC. Basic Protocol: Automated long-read genome finishing with short reads Support Protocol: Selecting optimal values for k-mer lengths (k) and Bloom filter size (b).
高质量的基因组组装对于许多生物学研究至关重要,而利用长测序读长可以帮助提高组装连续性。虽然长读长可以解决基因组中复杂和重复的区域,但它们相对较高的相关错误率仍然是一个主要限制。长读长通常生成具有较低碱基质量的草图基因组组装,必须通过基因组抛光步骤进行校正。混合基因组抛光解决方案可以通过利用更准确的短读长来验证碱基并纠正错误,从而大大提高长读长基因组组装的质量。目前可用的混合抛光方法依赖于读长比对,因此内存密集,并且不能很好地扩展到大型基因组。在这里,我们描述了 ntEdit+Sealer,这是一种无比对、基于 k-mer 的基因组完成协议,它采用了内存高效的布隆过滤器。该协议包括用于校正碱基错误和小插入缺失、标记潜在问题区域的 ntEdit,以及用于填充组装缺口和 ntEdit 标记的问题区域的 Sealer。ntEdit+Sealer 生成高度准确、纠错的基因组组装,可从 https://github.com/bcgsc/ntedit_sealer_protocol 获得 Makefile 管道。© 2022 作者。Wiley Periodicals LLC 出版的《当代协议》。基本方案:使用短读长自动进行长读长基因组完成 支持方案:选择最佳的 k-mer 长度(k)和布隆过滤器大小(b)值。