Canada's Michael Smith Genome Sciences Centre, BC Cancer Research, 100-570 West 7th Avenue, Vancouver, BC, V5Z 4S6, Canada.
BMC Bioinformatics. 2021 Oct 30;22(1):534. doi: 10.1186/s12859-021-04451-7.
Generating high-quality de novo genome assemblies is foundational to the genomics study of model and non-model organisms. In recent years, long-read sequencing has greatly benefited genome assembly and scaffolding, a process by which assembled sequences are ordered and oriented through the use of long-range information. Long reads are better able to span repetitive genomic regions compared to short reads, and thus have tremendous utility for resolving problematic regions and helping generate more complete draft assemblies. Here, we present LongStitch, a scalable pipeline that corrects and scaffolds draft genome assemblies exclusively using long reads.
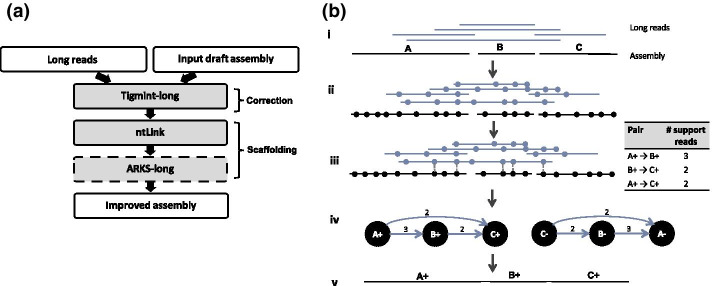
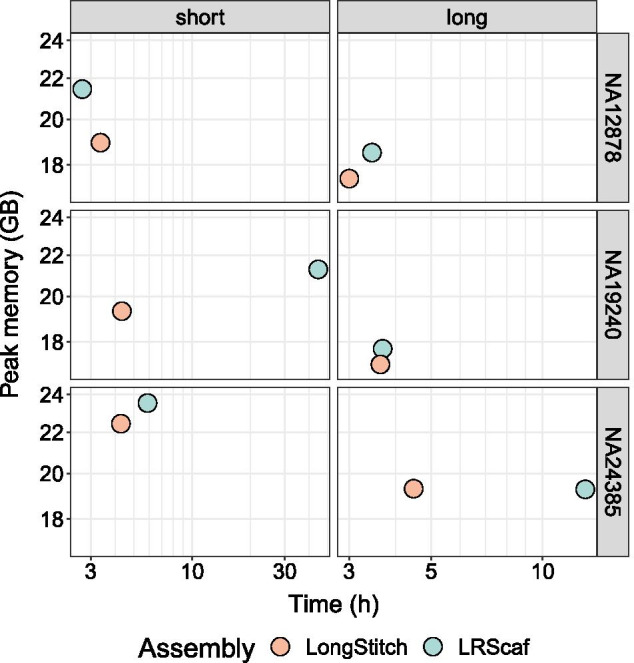
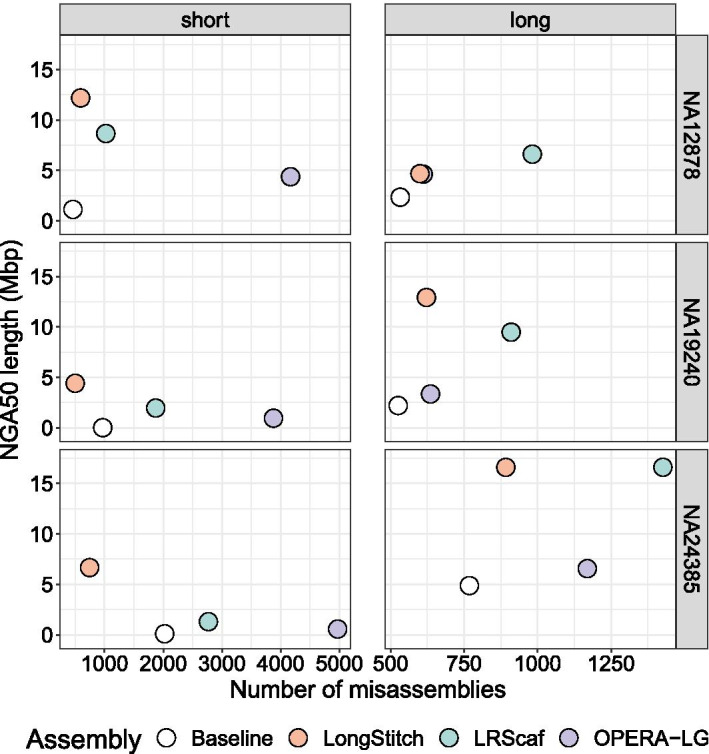
LongStitch incorporates multiple tools developed by our group and runs in up to three stages, which includes initial assembly correction (Tigmint-long), followed by two incremental scaffolding stages (ntLink and ARKS-long). Tigmint-long and ARKS-long are misassembly correction and scaffolding utilities, respectively, previously developed for linked reads, that we adapted for long reads. Here, we describe the LongStitch pipeline and introduce our new long-read scaffolder, ntLink, which utilizes lightweight minimizer mappings to join contigs. LongStitch was tested on short and long-read assemblies of Caenorhabditis elegans, Oryza sativa, and three different human individuals using corresponding nanopore long-read data, and improves the contiguity of each assembly from 1.2-fold up to 304.6-fold (as measured by NGA50 length). Furthermore, LongStitch generates more contiguous and correct assemblies compared to state-of-the-art long-read scaffolder LRScaf in most tests, and consistently improves upon human assemblies in under five hours using less than 23 GB of RAM.
Due to its effectiveness and efficiency in improving draft assemblies using long reads, we expect LongStitch to benefit a wide variety of de novo genome assembly projects. The LongStitch pipeline is freely available at https://github.com/bcgsc/longstitch .
生成高质量的从头基因组组装是模型和非模型生物基因组学研究的基础。近年来,长读测序极大地促进了基因组组装和支架的构建,该过程通过使用长程信息对组装序列进行排序和定向。与短读相比,长读能够更好地跨越重复基因组区域,因此对于解决有问题的区域并帮助生成更完整的草图组装具有巨大的实用价值。在这里,我们展示了 LongStitch,这是一个仅使用长读来纠正和支架草图基因组组装的可扩展流水线。
LongStitch 整合了我们小组开发的多个工具,最多可以分三个阶段运行,包括初始组装纠正(Tigmint-long),然后是两个增量支架阶段(ntLink 和 ARKS-long)。Tigmint-long 和 ARKS-long 分别是我们为链接读开发的错误组装纠正和支架工具,我们对其进行了调整以适应长读。在这里,我们描述了 LongStitch 流水线,并介绍了我们的新长读支架 ntLink,它利用轻量级的 minimizer 映射来连接 contigs。使用相应的纳米孔长读数据,对秀丽隐杆线虫、水稻和三个不同个体的短读和长读组装进行了 LongStitch 测试,每个组装的连续性都提高了 1.2 倍到 304.6 倍(以 NGA50 长度衡量)。此外,在大多数测试中,与最先进的长读支架 LRScaf 相比,LongStitch 生成了更连续和正确的组装,并且在不到五个小时的时间内使用不到 23GB 的 RAM 始终可以提高人类组装的质量。
由于其使用长读来有效提高草图组装的效果和效率,我们预计 LongStitch 将使各种从头基因组组装项目受益。LongStitch 流水线可在 https://github.com/bcgsc/longstitch 免费获得。