Zhou Jieshang, Yuan Yali, Xu Pengli, Yi Bin, Chen Hongju, Su Wei
People's Hospital of Dingxi City, The Second Affiliated Hospital of Lanzhou University Medical School, Dingxi, China.
College of Clinical Medicine, Lanzhou University Medical School, Lanzhou, China.
Evol Bioinform Online. 2022 May 13;18:11769343221095858. doi: 10.1177/11769343221095858. eCollection 2022.
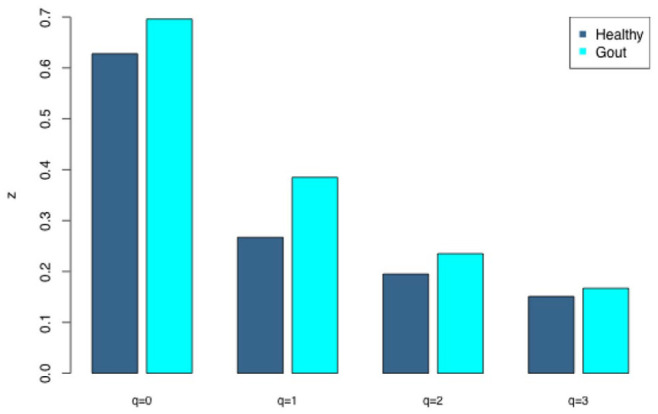
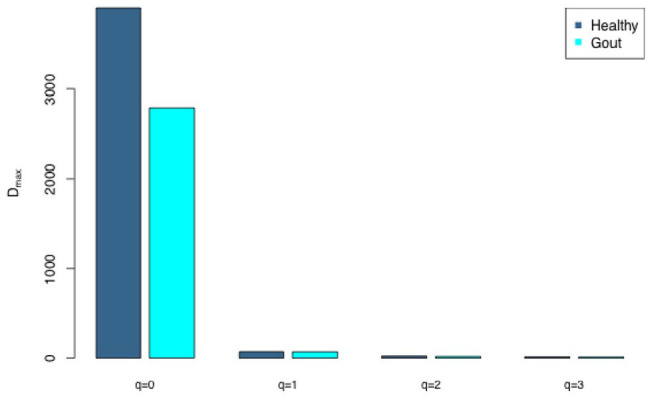
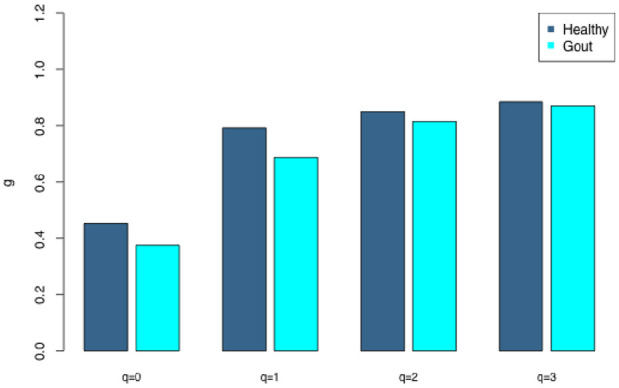
Gout is a prevalent chronic inflammatory disease that affects the life of tens of millions of people worldwide, and it typically presents as gout arthritis, gout stone, or even kidney damage. Research has revealed its connection with the gut microbiome, although exact mechanism is still unclear. Studies have shown the decline of microbiome diversity in gout patients and change of microbiome compositions between the gout patients and healthy controls. Nevertheless, how diversity changes across host individuals at a cohort () level has not been investigated to the best of our knowledge. Here we apply the diversity-area relationship (DAR), which is an extension to the classic SAR (species-area relationship) and establishes the power-function model between microbiome diversity and the number of individuals within cohort, to comparatively investigate diversity scaling (changes) of gut microbiome in gout patients and healthy controls. The DAR modeling with a study involving 83 subjects (41 gout patients) revealed that the potential number of microbial species in gout patients is only 70% of that in the healthy control (2790 vs 3900) although the difference may not be statistically significant. The other DAR parameters including diversity scaling and similarity parameters did not show statistically significant differences. We postulate that the high resilience of gut microbiome may explain the lack of significant gout-disease effects on gut microbial diversity at the population level. The lack of statistically significant difference between the gout patients and healthy controls at host population (cohort) level is different from the previous findings at individual level in the existing literature.
痛风是一种常见的慢性炎症性疾病,影响着全球数千万人的生活,通常表现为痛风性关节炎、痛风石,甚至肾脏损害。研究已经揭示了它与肠道微生物群的联系,尽管确切机制仍不清楚。研究表明痛风患者的微生物群多样性下降,且痛风患者与健康对照之间的微生物群组成存在变化。然而,据我们所知,尚未在队列水平上研究宿主个体间多样性是如何变化的。在此,我们应用多样性-面积关系(DAR),它是经典物种-面积关系(SAR)的扩展,建立了微生物群多样性与队列中个体数量之间的幂函数模型,以比较研究痛风患者和健康对照中肠道微生物群的多样性缩放(变化)。对一项涉及83名受试者(41名痛风患者)的研究进行DAR建模发现,痛风患者中潜在的微生物物种数量仅为健康对照的70%(2790对3900),尽管差异可能无统计学意义。其他DAR参数,包括多样性缩放和相似性参数,均未显示出统计学上的显著差异。我们推测,肠道微生物群的高弹性可能解释了在群体水平上痛风疾病对肠道微生物多样性缺乏显著影响的原因。痛风患者与健康对照在宿主群体(队列)水平上缺乏统计学上的显著差异,这与现有文献中个体水平的先前发现不同。