Program in Molecular Medicine, UMASS Chan Medical School, Worcester, MA 01605, USA.
Lewis-Sigler Institute for Quantitative Genomics, Princeton University, Princeton, NJ 08540, USA.
Genetics. 2022 Jul 30;221(4). doi: 10.1093/genetics/iyac085.
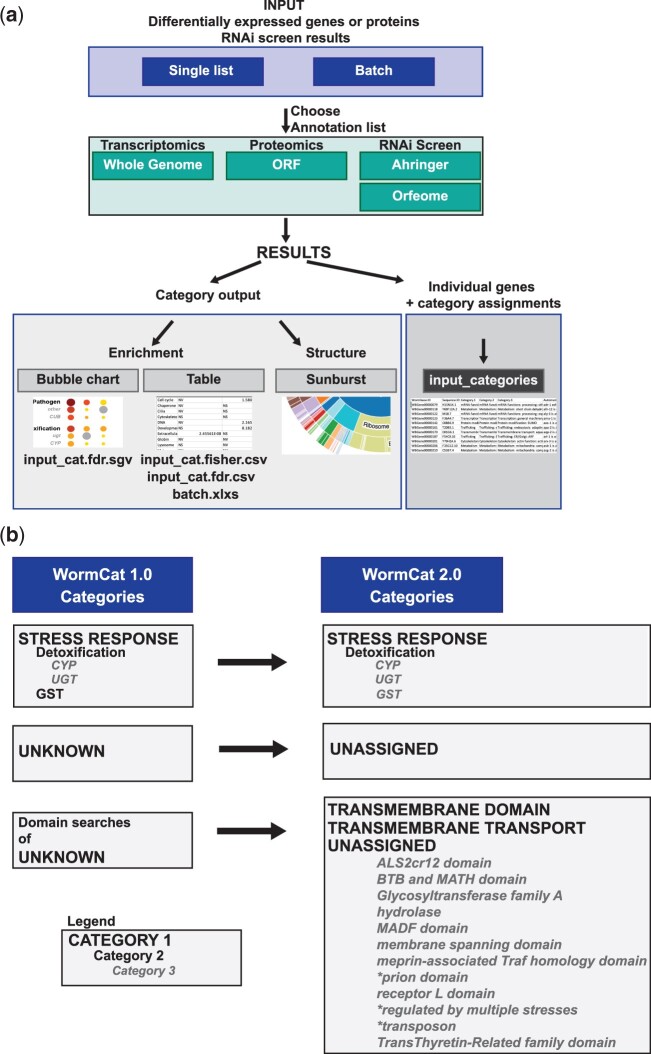
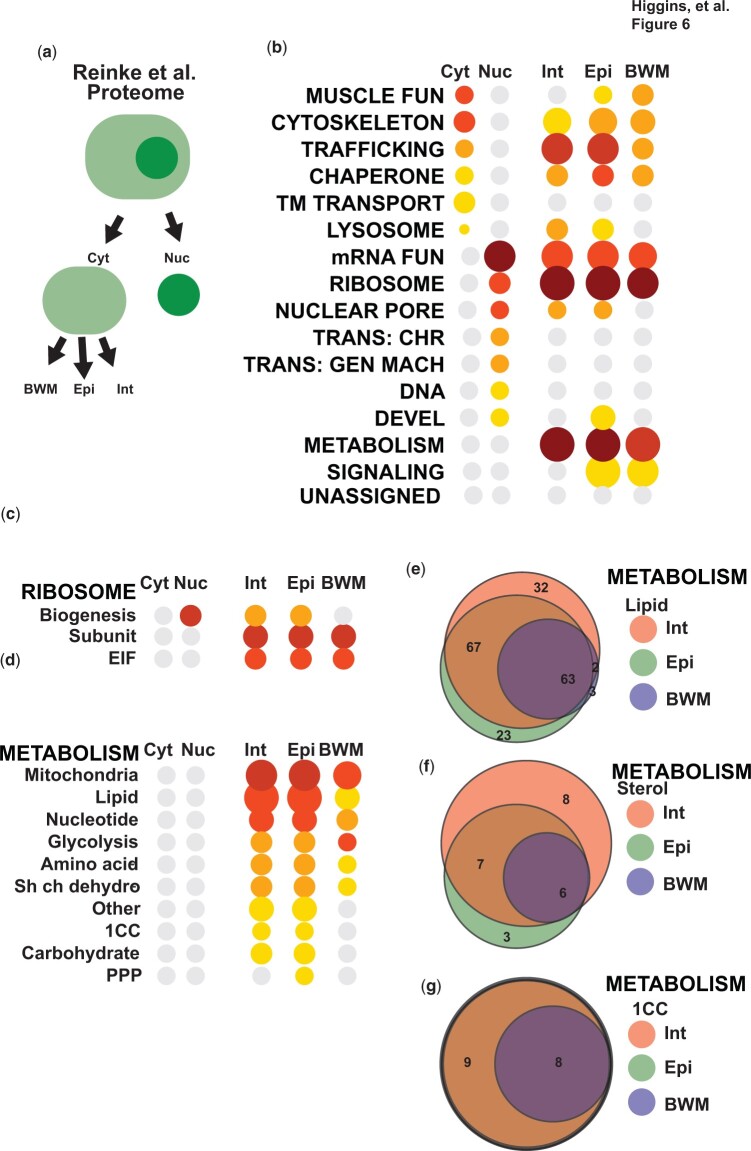
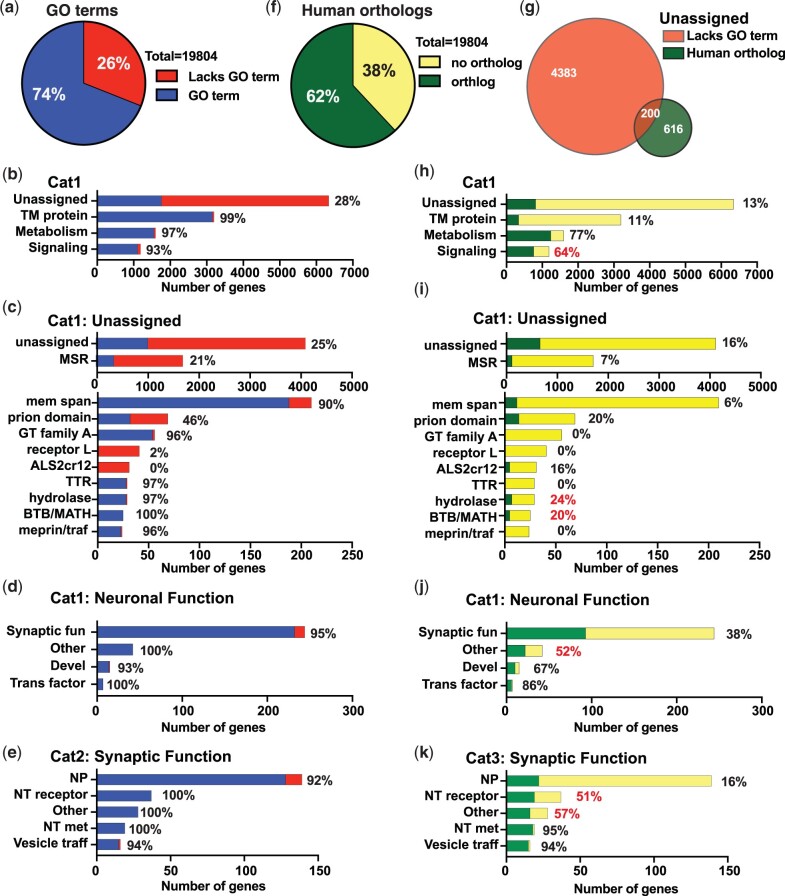
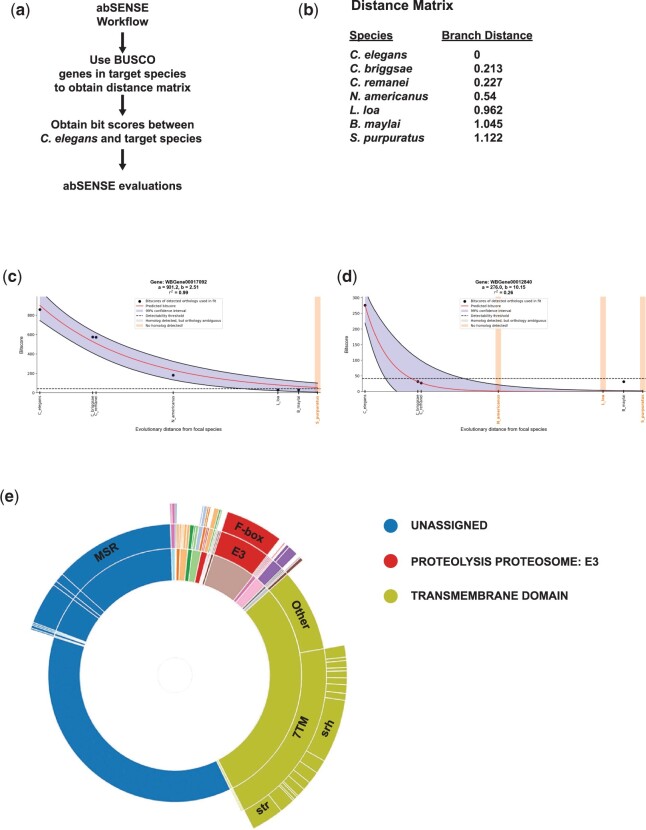
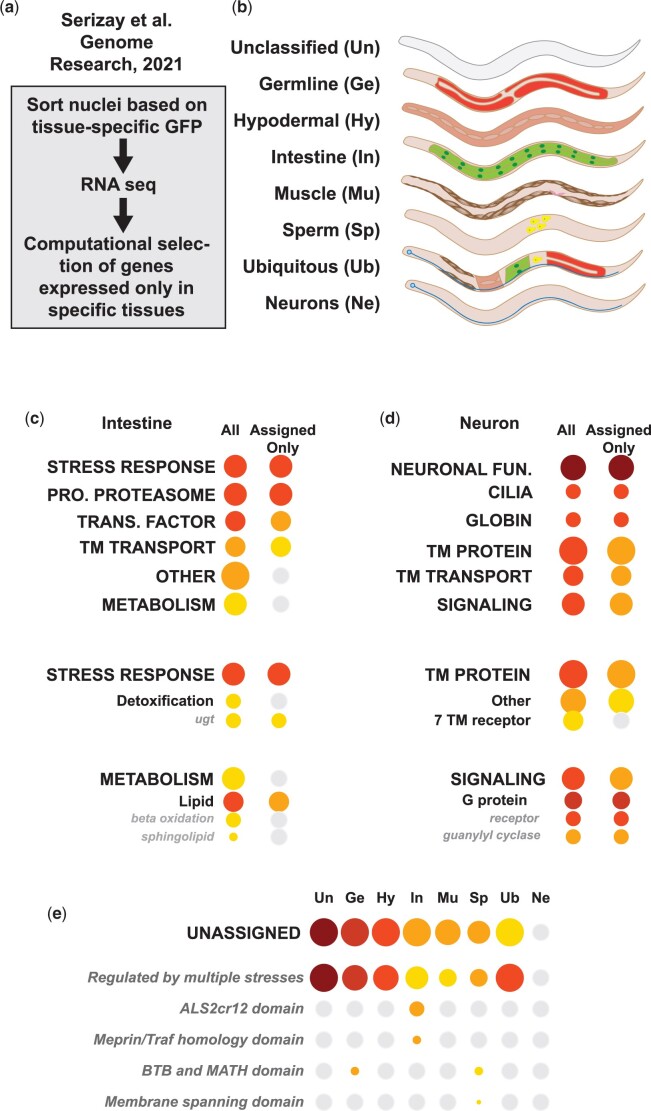
Omics tools provide broad datasets for biological discovery. However, the computational tools for identifying important genes or pathways in RNA-seq, proteomics, or GWAS (Genome-Wide Association Study) data depend on Gene Ontogeny annotations and are biased toward well-described pathways. This limits their utility as poorly annotated genes, which could have novel functions, are often passed over. Recently, we developed an annotation and category enrichment tool for Caenorhabditis elegans genomic data, WormCat, which provides an intuitive visualization output. Unlike Gene Ontogeny-based enrichment tools, which exclude genes with no annotation information, WormCat 2.0 retains these genes as a special UNASSIGNED category. Here, we show that the UNASSIGNED gene category enrichment exhibits tissue-specific expression patterns and can include genes with biological functions identified in published datasets. Poorly annotated genes are often considered to be potentially species-specific and thus, of reduced interest to the biomedical community. Instead, we find that around 3% of the UNASSIGNED genes have human orthologs, including some linked to human diseases. These human orthologs themselves have little annotation information. A recently developed method that incorporates lineage relationships (abSENSE) indicates that the failure of BLAST to detect homology explains the apparent lineage specificity for many UNASSIGNED genes. This suggests that a larger subset could be related to human genes. WormCat provides an annotation strategy that allows the association of UNASSIGNED genes with specific phenotypes and known pathways. Building these associations in C. elegans, with its robust genetic tools, provides a path to further functional study and insight into these understudied genes.
组学工具为生物发现提供了广泛的数据集。然而,在 RNA-seq、蛋白质组学或 GWAS(全基因组关联研究)数据中识别重要基因或途径的计算工具依赖于基因本体注释,并且偏向于描述良好的途径。这限制了它们的实用性,因为注释不良的基因可能具有新的功能,往往被忽视。最近,我们开发了一种用于秀丽隐杆线虫基因组数据的注释和类别富集工具 WormCat,它提供了直观的可视化输出。与基于基因本体的富集工具不同,后者排除了没有注释信息的基因,WormCat 2.0 将这些基因保留为特殊的“未分配”类别。在这里,我们表明“未分配”基因类别的富集表现出组织特异性表达模式,并且可以包括在已发表数据集中确定的具有生物学功能的基因。注释不良的基因通常被认为是潜在的物种特异性的,因此对生物医学社区的兴趣降低。相反,我们发现大约 3%的“未分配”基因具有人类同源基因,其中一些与人类疾病有关。这些人类同源基因本身的注释信息很少。最近开发的一种整合谱系关系的方法(abSENSE)表明,BLAST 未能检测到同源性解释了许多“未分配”基因明显的谱系特异性。这表明更大的子集可能与人类基因有关。WormCat 提供了一种注释策略,允许将“未分配”基因与特定表型和已知途径相关联。在秀丽隐杆线虫中建立这些关联,利用其强大的遗传工具,为进一步研究这些研究不足的基因的功能和深入了解提供了途径。