Welcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, United Kingdom.
PLoS One. 2011;6(8):e23216. doi: 10.1371/journal.pone.0023216. Epub 2011 Aug 15.
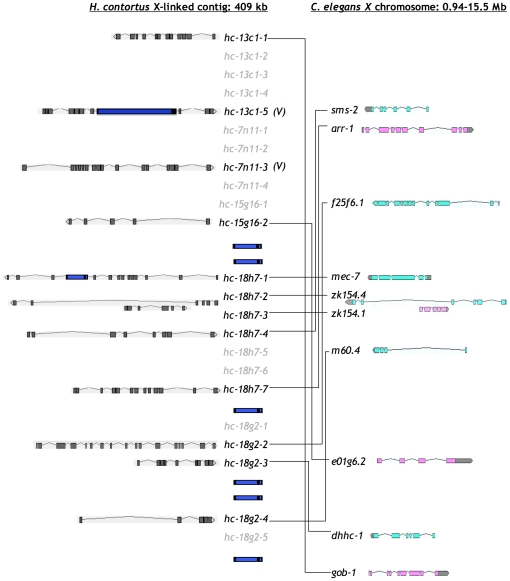
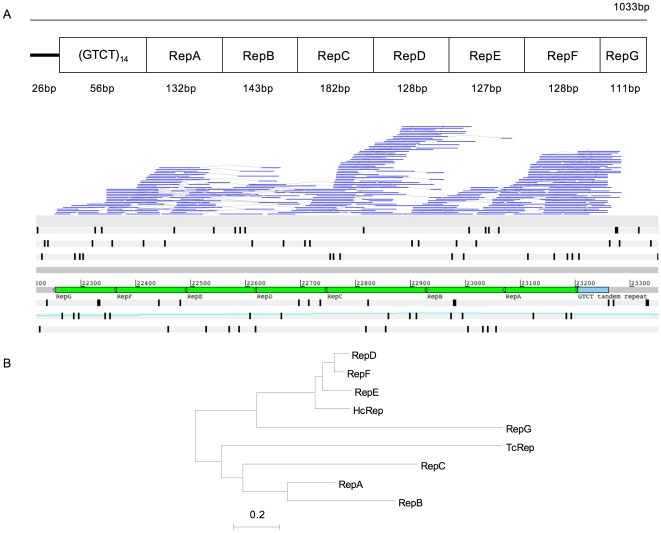
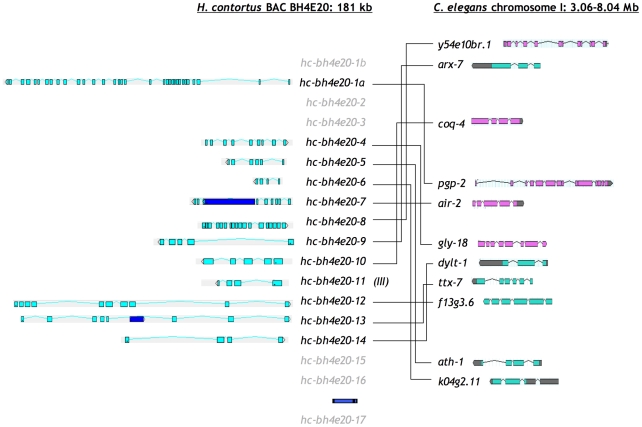
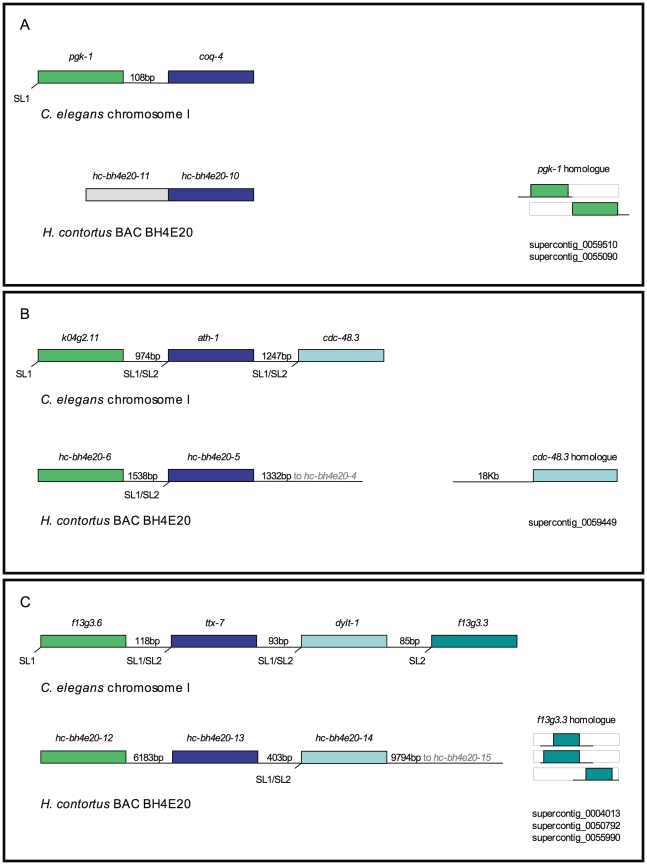
The genomes of numerous parasitic nematodes are currently being sequenced, but their complexity and size, together with high levels of intra-specific sequence variation and a lack of reference genomes, makes their assembly and annotation a challenging task. Haemonchus contortus is an economically significant parasite of livestock that is widely used for basic research as well as for vaccine development and drug discovery. It is one of many medically and economically important parasites within the strongylid nematode group. This group of parasites has the closest phylogenetic relationship with the model organism Caenorhabditis elegans, making comparative analysis a potentially powerful tool for genome annotation and functional studies. To investigate this hypothesis, we sequenced two contiguous fragments from the H. contortus genome and undertook detailed annotation and comparative analysis with C. elegans. The adult H. contortus transcriptome was sequenced using an Illumina platform and RNA-seq was used to annotate a 409 kb overlapping BAC tiling path relating to the X chromosome and a 181 kb BAC insert relating to chromosome I. In total, 40 genes and 12 putative transposable elements were identified. 97.5% of the annotated genes had detectable homologues in C. elegans of which 60% had putative orthologues, significantly higher than previous analyses based on EST analysis. Gene density appears to be less in H. contortus than in C. elegans, with annotated H. contortus genes being an average of two-to-three times larger than their putative C. elegans orthologues due to a greater intron number and size. Synteny appears high but gene order is generally poorly conserved, although areas of conserved microsynteny are apparent. C. elegans operons appear to be partially conserved in H. contortus. Our findings suggest that a combination of RNA-seq and comparative analysis with C. elegans is a powerful approach for the annotation and analysis of strongylid nematode genomes.
目前正在对大量寄生线虫的基因组进行测序,但由于其复杂性和大小、种内序列变异水平高以及缺乏参考基因组,使得它们的组装和注释成为一项具有挑战性的任务。旋毛虫是一种对家畜具有重要经济意义的寄生虫,广泛用于基础研究以及疫苗开发和药物发现。它是强杆线虫组中许多具有医学和经济重要性的寄生虫之一。该寄生虫组与模式生物秀丽隐杆线虫具有最密切的系统发育关系,使得比较分析成为基因组注释和功能研究的一种潜在强大工具。为了研究这一假说,我们对旋毛虫基因组的两个连续片段进行了测序,并与秀丽隐杆线虫进行了详细的注释和比较分析。使用 Illumina 平台对旋毛虫成虫转录组进行了测序,并使用 RNA-seq 对涉及 X 染色体的 409 kb 重叠 BAC 平铺路径和涉及染色体 I 的 181 kb BAC 插入物进行了注释。总共鉴定了 40 个基因和 12 个推定转座元件。在可检测到秀丽隐杆线虫同源物的注释基因中,97.5%有假定的直系同源物,明显高于先前基于 EST 分析的分析。基因密度似乎低于秀丽隐杆线虫,注释的旋毛虫基因由于内含子数量和大小更大,平均是其假定的秀丽隐杆线虫直系同源物的两到三倍。同线性似乎很高,但基因顺序通常保存不佳,尽管存在明显的保守微同线性区域。秀丽隐杆线虫操纵子似乎在旋毛虫中部分保守。我们的研究结果表明,RNA-seq 与秀丽隐杆线虫的比较分析相结合是注释和分析强杆线虫基因组的一种强大方法。