van Santen Selveta Sanne, Daly Adrian F, Buchfelder Michael, Coras Roland, Zhao Yining, Beckers Albert, van der Lely Aart Jan, Hofland Leo J, Balvers Rutger K, van Koetsveld P, van den Heuvel-Eibrink Marry Marrigje, Neggers Sebastian Johannes Cornelis Martinus Maria
Department of Internal Medicine, Endocrinology; Erasmus Medical Center, Rotterdam, The Netherlands.
Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands.
AACE Clin Case Rep. 2021 Dec 16;8(3):119-123. doi: 10.1016/j.aace.2021.12.003. eCollection 2022 May-Jun.
Our objective was to describe the clinical course and treatment challenges in a very young patient with a pituitary adenoma due to a novel gene mutation, highlighting the limitations of somatostatin receptor immunohistochemistry to predict clinical responses to somatostatin analogs in acromegaly.
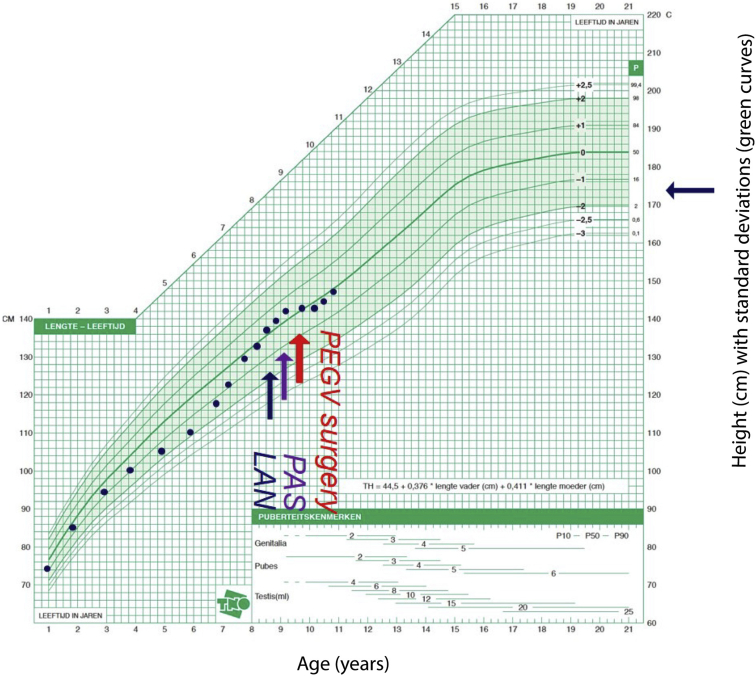
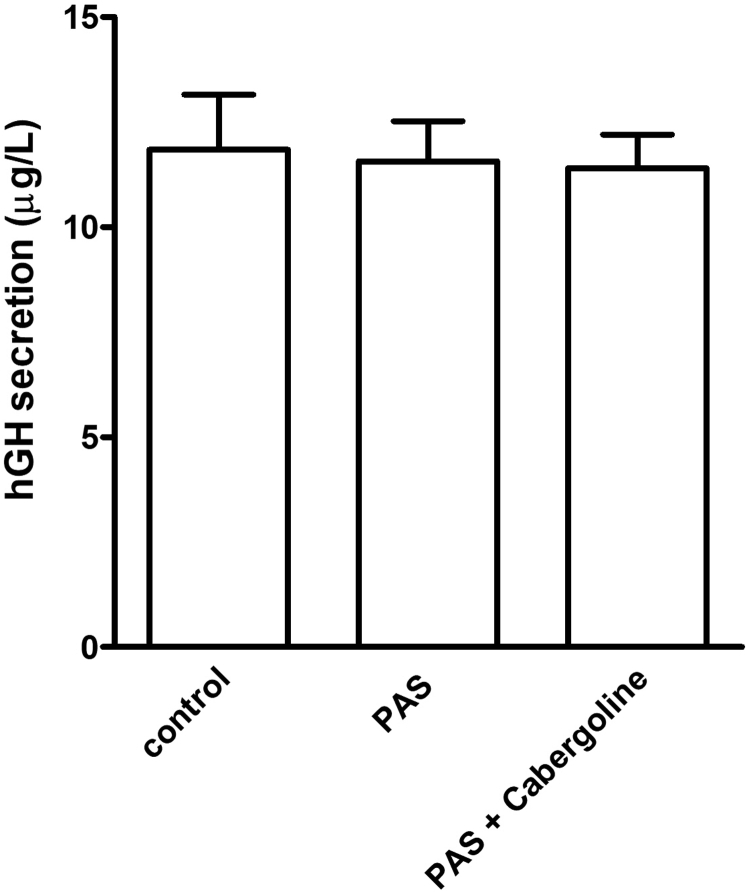
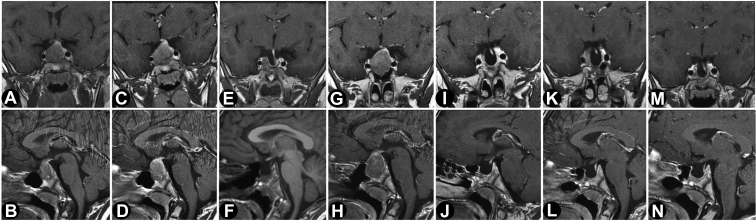
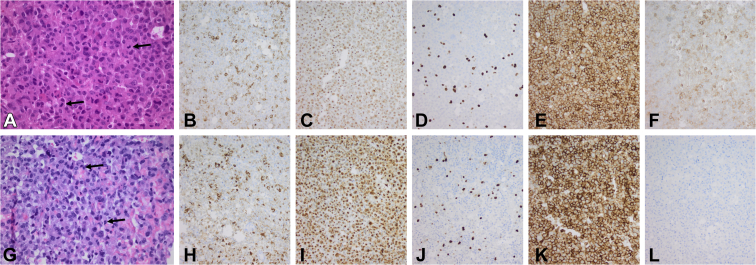
We report the case of a 7-year-old boy presenting with headache, visual field defects, and accelerated growth following failure to thrive. The laboratory results showed high insulin-like growth factor I (IGF-I) (standardised deviation scores ( +3.49) and prolactin levels (0.5 nmol/L), and magnetic resonance imaging identified a pituitary macroadenoma. Tumoral/hormonal control could not be achieved despite 3 neurosurgical procedures, each time with apparent total resection or with lanreotide or pasireotide. IGF-I levels decreased with the GH receptor antagonist pegvisomant. The loss of somatostatin receptor 5 was observed between the second and third tumor resection. In vitro, no effect on tumoral GH release by pasireotide (with/without cabergoline) was observed. Genetic analysis revealed a novel germline mutation: p.Tyr202∗ (pathogenic; class 4).
In vitro response of tumor tissue to somatostatin may better predict tumoral in vivo responses of somatostatin analogs than somatostatin receptor immunohistochemistry.
We identified a novel pathologic mutation that was associated with incipient acrogigantism in an extremely young patient who had a complicated course of disease. Growth acceleration can be masked due to failure to thrive. Tumoral growth hormone release in vivo may be predicted with in vitro exposure to somatostatin receptor analogs, as it cannot be assumed that all -mutated somatotropinomas respond well to pasireotide.
我们的目的是描述一名因新的基因突变而患有垂体腺瘤的极年轻患者的临床病程及治疗挑战,强调生长抑素受体免疫组化在预测肢端肥大症患者对生长抑素类似物临床反应方面的局限性。
我们报告了一名7岁男孩的病例,该男孩因发育迟缓后出现头痛、视野缺损和生长加速。实验室检查结果显示胰岛素样生长因子I(IGF-I)水平升高(标准化偏差评分(+3.49))和催乳素水平升高(0.5 nmol/L),磁共振成像显示垂体大腺瘤。尽管进行了3次神经外科手术,每次均为明显的全切手术或使用兰瑞肽或帕瑞肽,但仍未实现肿瘤/激素控制。使用生长激素受体拮抗剂培维索孟后IGF-I水平下降。在第二次和第三次肿瘤切除之间观察到生长抑素受体5缺失。在体外,未观察到帕瑞肽(加/不加卡麦角林)对肿瘤生长激素释放有影响。基因分析发现一种新的种系突变:p.Tyr202∗(致病性;4类)。
肿瘤组织对生长抑素的体外反应可能比生长抑素受体免疫组化更好地预测生长抑素类似物的肿瘤体内反应。
我们在一名患有复杂病程的极年轻患者中鉴定出一种与早期肢端肥大症相关的新的病理突变。生长加速可能因发育迟缓而被掩盖。由于不能假定所有突变的生长激素瘤都对帕瑞肽反应良好,因此可以通过体外暴露于生长抑素受体类似物来预测肿瘤体内生长激素的释放。