Department of Neurology, H. Houston Merritt Neuromuscular Research Center, Columbia University Irving Medical Center, New York, New York, USA.
Genetics Division, Department of Pediatrics, King Saud bin Abdulaziz University for Health Science, King Abdulaziz Medical City, Riyadh, Saudi Arabia.
J Clin Invest. 2022 Jul 1;132(13). doi: 10.1172/JCI145660.
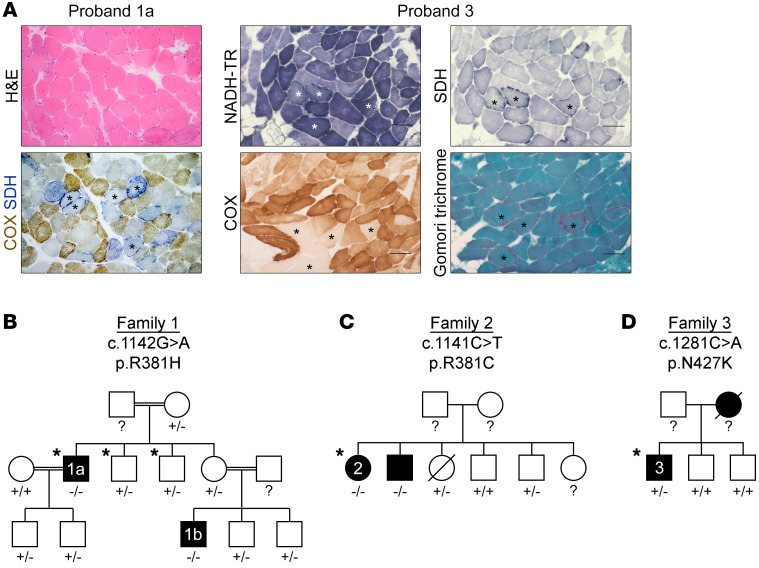
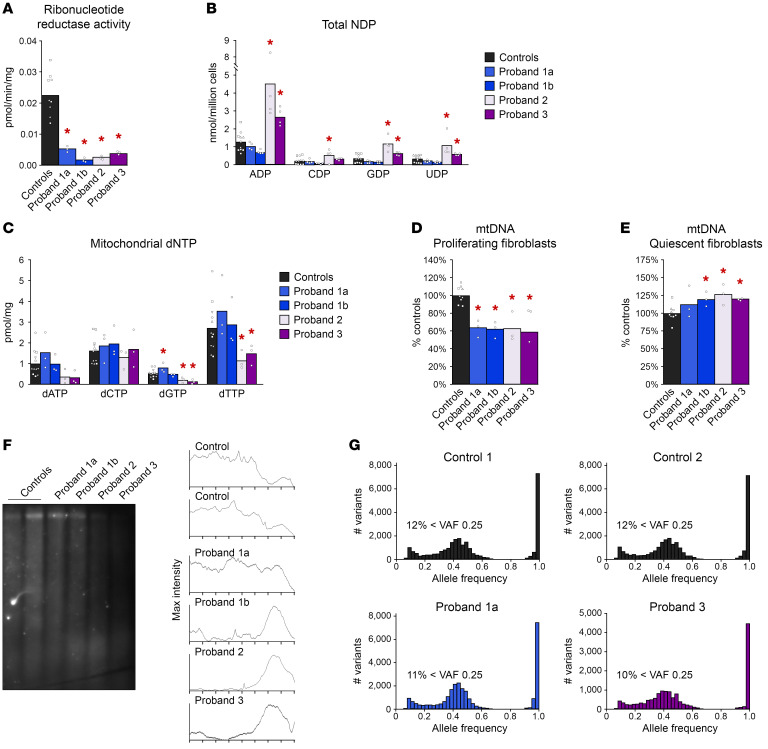
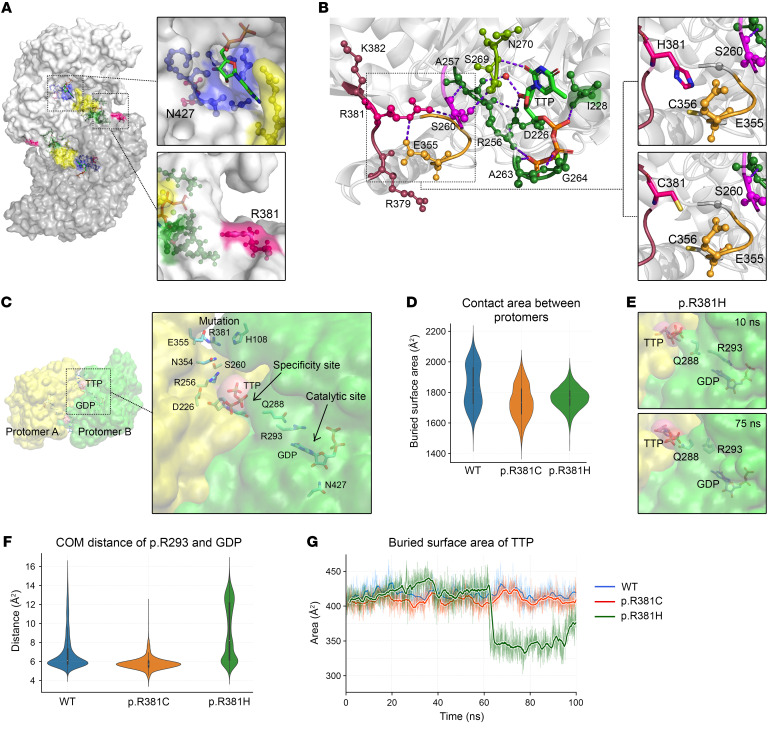
Mitochondrial DNA (mtDNA) depletion/deletions syndromes (MDDS) encompass a clinically and etiologically heterogenous group of mitochondrial disorders caused by impaired mtDNA maintenance. Among the most frequent causes of MDDS are defects in nucleoside/nucleotide metabolism, which is critical for synthesis and homeostasis of the deoxynucleoside triphosphate (dNTP) substrates of mtDNA replication. A central enzyme for generating dNTPs is ribonucleotide reductase, a critical mediator of de novo nucleotide synthesis composed of catalytic RRM1 subunits in complex with RRM2 or p53R2. Here, we report 5 probands from 4 families who presented with ptosis and ophthalmoplegia as well as other clinical manifestations and multiple mtDNA deletions in muscle. We identified 3 RRM1 loss-of-function variants, including a dominant catalytic site variant (NP_001024.1: p.N427K) and 2 homozygous recessive variants at p.R381, which has evolutionarily conserved interactions with the specificity site. Atomistic molecular dynamics simulations indicate mechanisms by which RRM1 variants affect protein structure. Cultured primary skin fibroblasts of probands manifested mtDNA depletion under cycling conditions, indicating impaired de novo nucleotide synthesis. Fibroblasts also exhibited aberrant nucleoside diphosphate and dNTP pools and mtDNA ribonucleotide incorporation. Our data reveal that primary RRM1 deficiency and, by extension, impaired de novo nucleotide synthesis are causes of MDDS.
线粒体 DNA(mtDNA)耗竭/缺失综合征(MDDS)包括一组由 mtDNA 维持受损引起的临床和病因学上异质性的线粒体疾病。MDDS 最常见的原因之一是核苷/核苷酸代谢缺陷,这对 mtDNA 复制的脱氧核苷三磷酸(dNTP)底物的合成和动态平衡至关重要。生成 dNTP 的核心酶是核核苷酸还原酶,它是从头核苷酸合成的关键介质,由与 RRM2 或 p53R2 复合的催化 RRM1 亚基组成。在这里,我们报告了 4 个家系中的 5 个先证者,他们表现为眼睑下垂和眼肌麻痹以及其他临床表现和肌肉中的多个 mtDNA 缺失。我们鉴定了 3 个 RRM1 功能丧失变异体,包括显性催化位点变异体(NP_001024.1:p.N427K)和 2 个同源隐性变异体 p.R381,它与特异性位点具有进化上保守的相互作用。原子分子动力学模拟表明了 RRM1 变异体影响蛋白质结构的机制。先证者的培养原代皮肤成纤维细胞在循环条件下表现出 mtDNA 耗竭,表明从头核苷酸合成受损。成纤维细胞还表现出异常的核苷二磷酸和 dNTP 池以及 mtDNA 核糖核苷酸掺入。我们的数据表明,原发性 RRM1 缺乏症,进而导致从头核苷酸合成受损,是 MDDS 的原因。