Division of Metabolism, Endocrinology & Diabetes, University of Michigan Medical Center, Ann Arbor, Michigan, USA.
Departments of Medicine, Pediatrics, and Committee on Genetics, The University of Chicago, Chicago Illinois, USA.
J Biol Chem. 2022 Jul;298(7):102066. doi: 10.1016/j.jbc.2022.102066. Epub 2022 May 23.
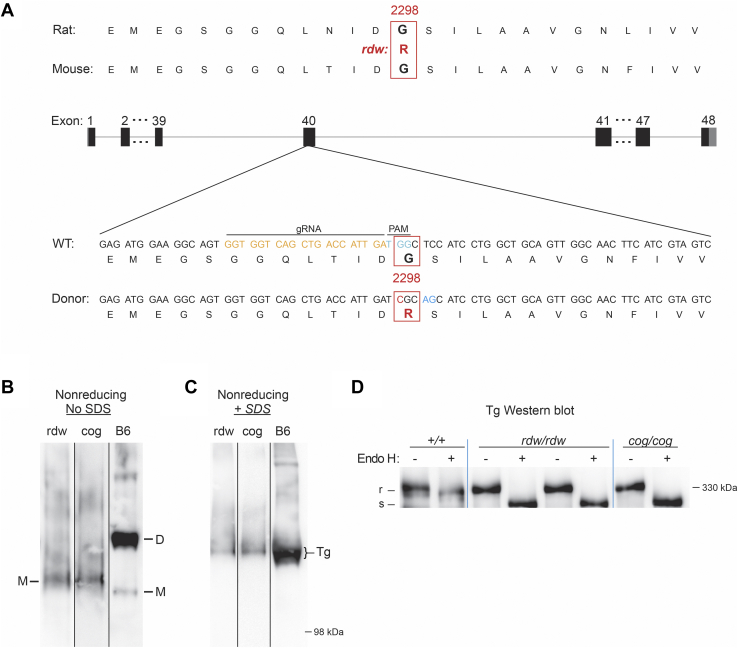
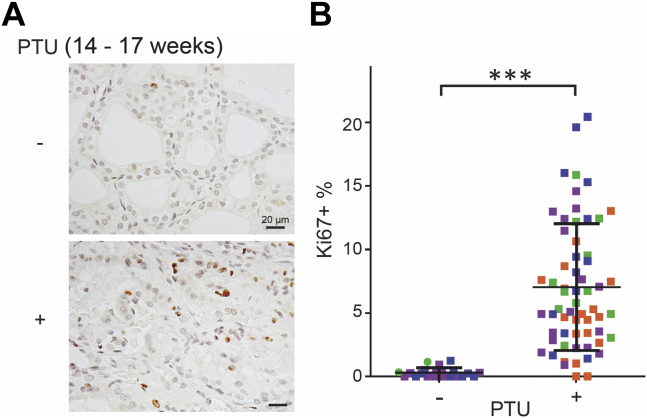
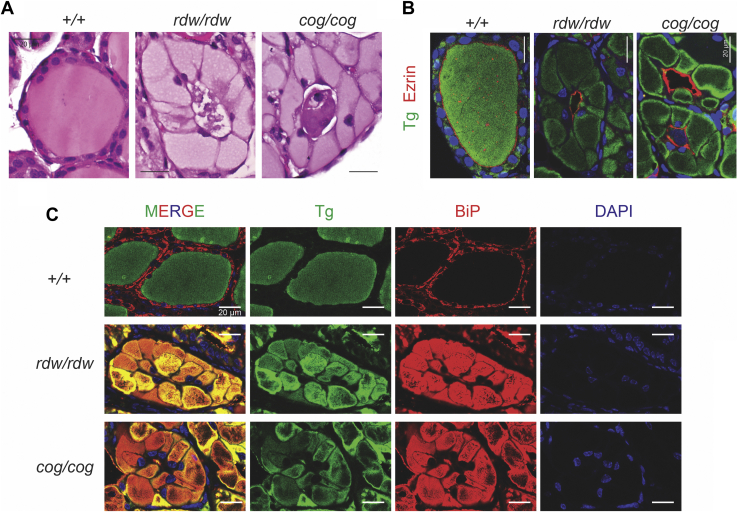
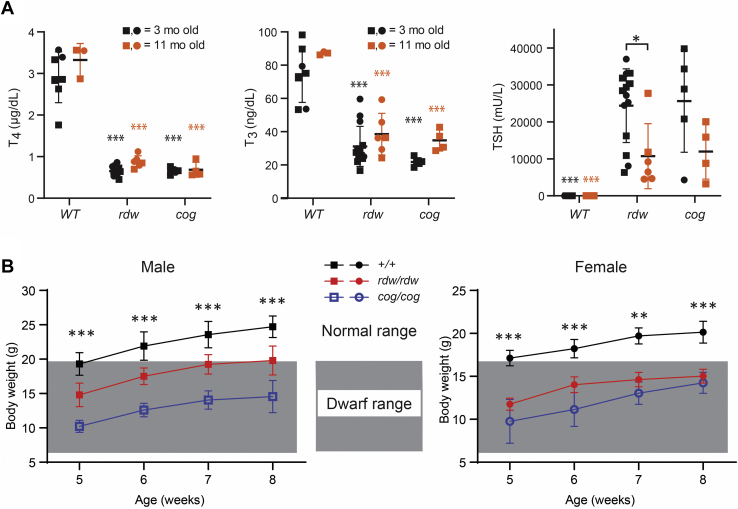
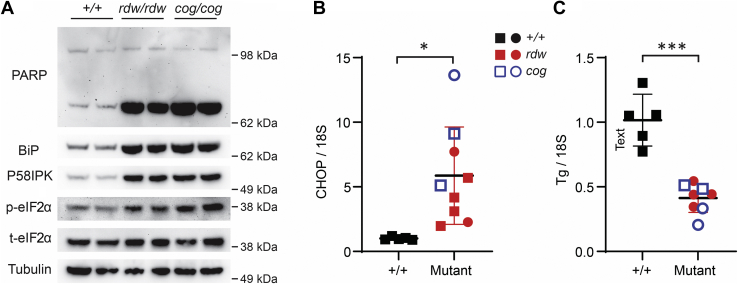
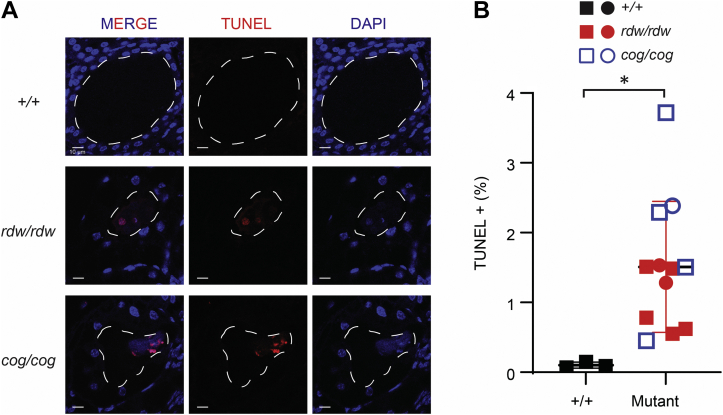
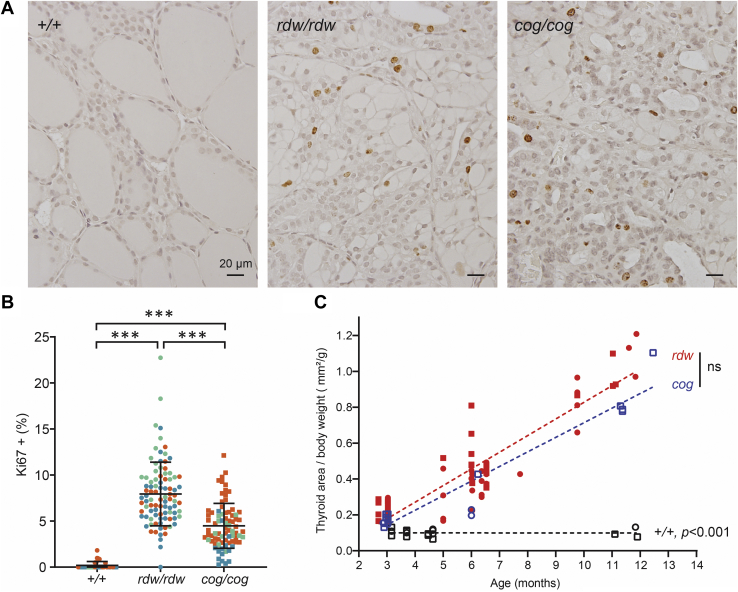
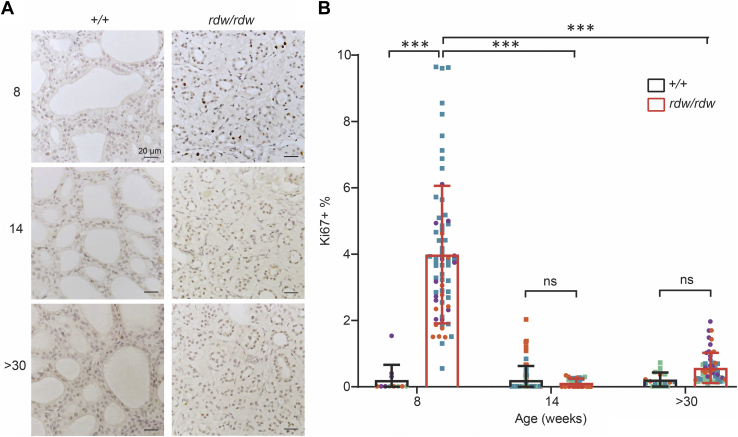
Congenital hypothyroidism with biallelic thyroglobulin (Tg protein, encoded by the TG gene) mutation is an endoplasmic reticulum (ER) storage disease. Many patients (and animal models) grow an enlarged thyroid (goiter), yet some do not. In adulthood, hypothyroid TG mice (bearing a Tg-L2263P mutation) exhibit a large goiter, whereas adult WIC rats bearing the TG mutation (Tg-G2298R) exhibit a hypoplastic thyroid. Homozygous TG mutation has been linked to thyroid cell death, and cytotoxicity of the Tg-G2298R protein was previously thought to explain the lack of goiter in WIC-TG rats. However, recent studies revealed that TG mice also exhibit widespread ER stress-mediated thyrocyte death, yet under continuous feedback stimulation, thyroid cells proliferate in excess of their demise. Here, to examine the relative proteotoxicity of the Tg-G2298R protein, we have used CRISPR-CRISPR-associated protein 9 technology to generate homozygous TG knock-in mice in a strain background identical to that of TG mice. TG mice exhibit similar phenotypes of defective Tg protein folding, thyroid histological abnormalities, hypothyroidism, and growth retardation. TG mice do not show evidence of greater ER stress response or stress-mediated cell death than TG mice, and both mouse models exhibit sustained thyrocyte proliferation, with comparable goiter growth. In contrast, in WIC-TG rats, as a function of aging, the thyrocyte proliferation rate declines precipitously. We conclude that the mutant Tg-G2298R protein is not intrinsically more proteotoxic than Tg-L2263P; rather, aging-dependent difference in maintenance of cell proliferation is the limiting factor, which accounts for the absence of goiter in adult WIC-TG rats.
先天性甲状腺功能减退症与甲状腺球蛋白(Tg 蛋白,由 TG 基因编码)双等位基因突变是一种内质网(ER)储存疾病。许多患者(和动物模型)甲状腺肿大(甲状腺肿),但有些则没有。在成年期,患有甲状腺功能减退症的 Tg 小鼠(携带 Tg-L2263P 突变)表现出巨大的甲状腺肿,而携带 TG 突变(Tg-G2298R)的成年 WIC 大鼠则表现出甲状腺发育不全。纯合 TG 突变与甲状腺细胞死亡有关,先前认为 Tg-G2298R 蛋白的细胞毒性解释了 WIC-TG 大鼠甲状腺肿的缺乏。然而,最近的研究表明,Tg 小鼠也表现出广泛的 ER 应激介导的甲状腺细胞死亡,尽管持续的反馈刺激,甲状腺细胞过度增殖超过其死亡。在这里,为了研究 Tg-G2298R 蛋白的相对蛋白毒性,我们使用 CRISPR-CRISPR 相关蛋白 9 技术在与 Tg 小鼠相同的品系背景下生成纯合 TG 基因敲入小鼠。Tg 小鼠表现出类似的 Tg 蛋白折叠缺陷、甲状腺组织学异常、甲状腺功能减退和生长迟缓的表型。Tg 小鼠没有表现出比 Tg 小鼠更大的 ER 应激反应或应激介导的细胞死亡的证据,并且两种小鼠模型都表现出持续的甲状腺细胞增殖,具有相似的甲状腺肿生长。相比之下,在 WIC-TG 大鼠中,随着年龄的增长,甲状腺细胞增殖率急剧下降。我们得出的结论是,突变的 Tg-G2298R 蛋白并不比 Tg-L2263P 更具内在的蛋白毒性;相反,与细胞增殖维持相关的衰老差异是限制因素,这解释了成年 WIC-TG 大鼠甲状腺肿缺失的原因。