Zárate-Potes Alejandra, Ali Irtiqa, Ribeiro Camacho Margarida, Brownless Hayley, Benedetto Alexandre
Division of Biomedical and Life Sciences, Lancaster University, Lancaster, United Kingdom.
Front Microbiol. 2022 May 10;13:853629. doi: 10.3389/fmicb.2022.853629. eCollection 2022.
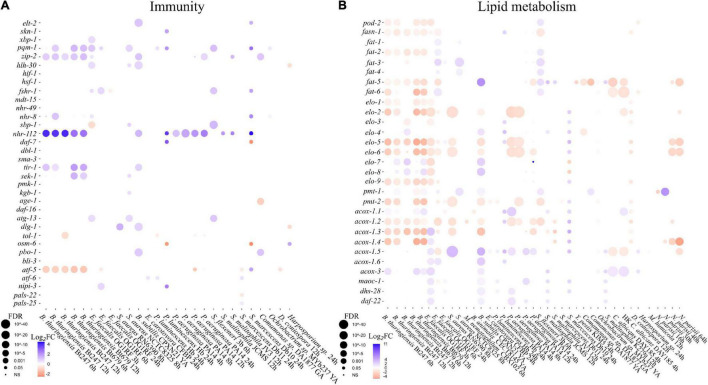
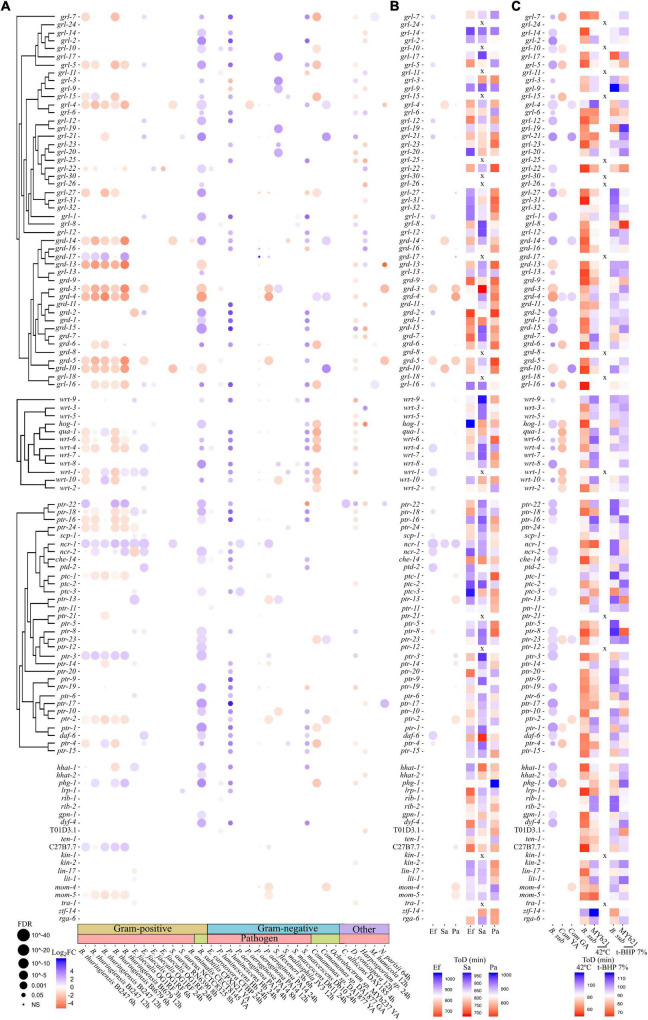
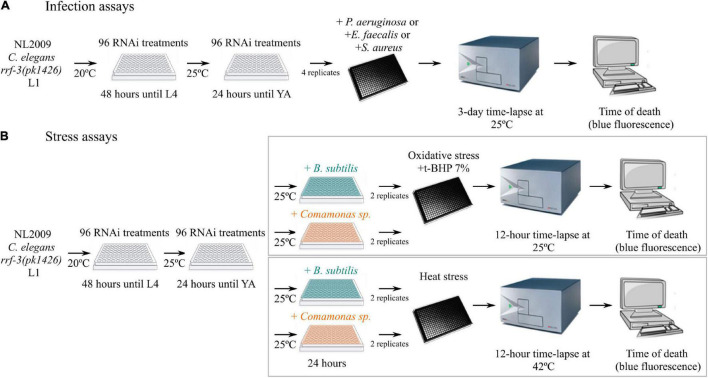
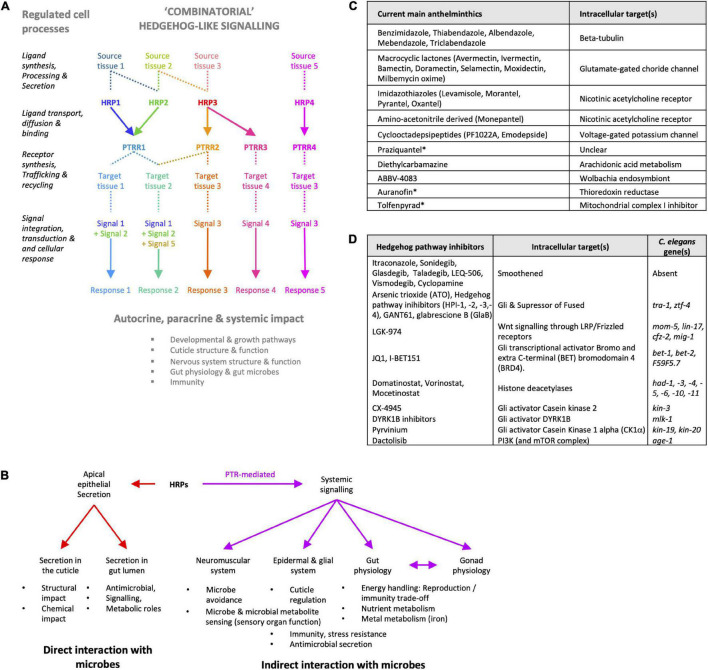
Controlling nematode-caused diseases that affect cattle and crops world-wide remains a critical economic issue, owing to the lack of effective sustainable interventions. The interdependence of roundworms and their environmental microbes, including their microbiota, offers an opportunity for developing more targeted anthelminthic strategies. However, paucity of information and a currently narrow understanding of nematode-microbe interactions limited to specific infection contexts has precluded us from exploiting it. With the advent of omics approaches to map host-microbe genetic interactions, particularly in the model roundworm , large datasets are now available across multiple models, that enable identification of nematode-microbe-specific pathways. In this work we collected 20 transcriptomic datasets documenting gene expression changes of exposed to 20 different commensal and pathogenic microbes, performing gene enrichment analyses followed by functional testing using RNA interference directed toward genes of interest, before contrasting results from transcriptomic meta-analyses and phenomics. Differential expression analyses revealed a broad enrichment in signaling, innate immune response and (lipid) metabolism genes. Amongst signaling gene families, the nematode-divergent and expanded Hedgehog-like signaling (HHLS) pathway featured prominently. Indeed, 24/60 Hedgehog-like proteins (HRPs) and 15/27 Patched-related receptors (PTRs) were differentially expressed in at least four microbial contexts, while up to 32/60 HRPs could be differentially expressed in a single context. interestingly, differentially expressed genes followed a microbe-specific pattern, suggestive of an adaptive microbe-specific response. To investigate this further, we knocked-down 96 individual HHLS genes by RNAi, using high-throughput assays to assess their impact on three worm-gut infection models (, , and ) and two worm-commensal paradigms ( sp., and ). We notably identified new putative infection response genes whose upregulation was required for normal pathogen resistance (i.e., and protective against ), as well as commensal-specific host-gene expression changes that are required for normal host stress handling. Importantly, interactions appeared more microbe-specific than shared. Our results thus implicate the Hedgehog-like signaling pathway in the modulation and possibly fine-tuning of nematode-microbe interactions and support the idea that interventions targeting this pathway may provide a new avenue for anthelmintic development.
由于缺乏有效的可持续干预措施,控制影响全球牛和农作物的线虫引起的疾病仍然是一个关键的经济问题。蛔虫与其环境微生物(包括其微生物群)的相互依存关系为开发更具针对性的驱虫策略提供了机会。然而,信息匮乏以及目前对线虫与微生物相互作用的理解局限于特定感染背景,这使得我们无法利用这一机会。随着组学方法的出现,用于绘制宿主与微生物的基因相互作用图谱,特别是在模式蛔虫中,现在多个模型都有大量数据集,这使得能够识别线虫与微生物特异性的途径。在这项工作中,我们收集了20个转录组数据集,记录了暴露于20种不同共生和致病微生物后的基因表达变化,进行基因富集分析,然后使用针对感兴趣基因的RNA干扰进行功能测试,最后对比转录组元分析和表型组学的结果。差异表达分析揭示了信号传导、先天免疫反应和(脂质)代谢基因的广泛富集。在信号基因家族中,线虫特异且扩展的类刺猬信号(HHLS)通路尤为突出。事实上,60种类刺猬蛋白(HRPs)中的24种和27种patched相关受体(PTRs)中的15种在至少四种微生物背景下差异表达,而在单一背景下多达32/60的HRPs可能差异表达。有趣的是,差异表达基因呈现出微生物特异性模式,提示存在适应性的微生物特异性反应。为了进一步研究这一点,我们通过RNA干扰敲低了96个单独的HHLS基因,使用高通量检测来评估它们对三种蠕虫肠道感染模型(分别为 、 和 )以及两种蠕虫共生模式( 种,和 )的影响。我们特别鉴定出了新的假定感染反应基因,其上调对于正常的病原体抗性是必需的(即 对 和 具有保护作用),以及正常宿主应激处理所需的共生特异性宿主基因表达变化。重要的是,相互作用似乎更具微生物特异性而非共享性。因此,我们的结果表明类刺猬信号通路参与了线虫与微生物相互作用的调节,甚至可能是微调,并支持这样一种观点,即针对该通路的干预措施可能为驱虫药物开发提供一条新途径。