Borel Valerie, Boeing Stefan, Van Wietmarschen Niek, Sridharan Sriram, Hill Bethany Rebekah, Ombrato Luigi, Perez-Lloret Jimena, Jackson Deb, Goldstone Robert, Boulton Simon J, Nussenzweig Andre, Bellelli Roberto
The Francis Crick Institute, 1 Midland Road, NW1 1AT London, UK.
Laboratory of Genome Integrity, National Cancer Institute, NIH, Bethesda, MD, USA.
Cell Rep. 2022 May 31;39(9):110871. doi: 10.1016/j.celrep.2022.110871.
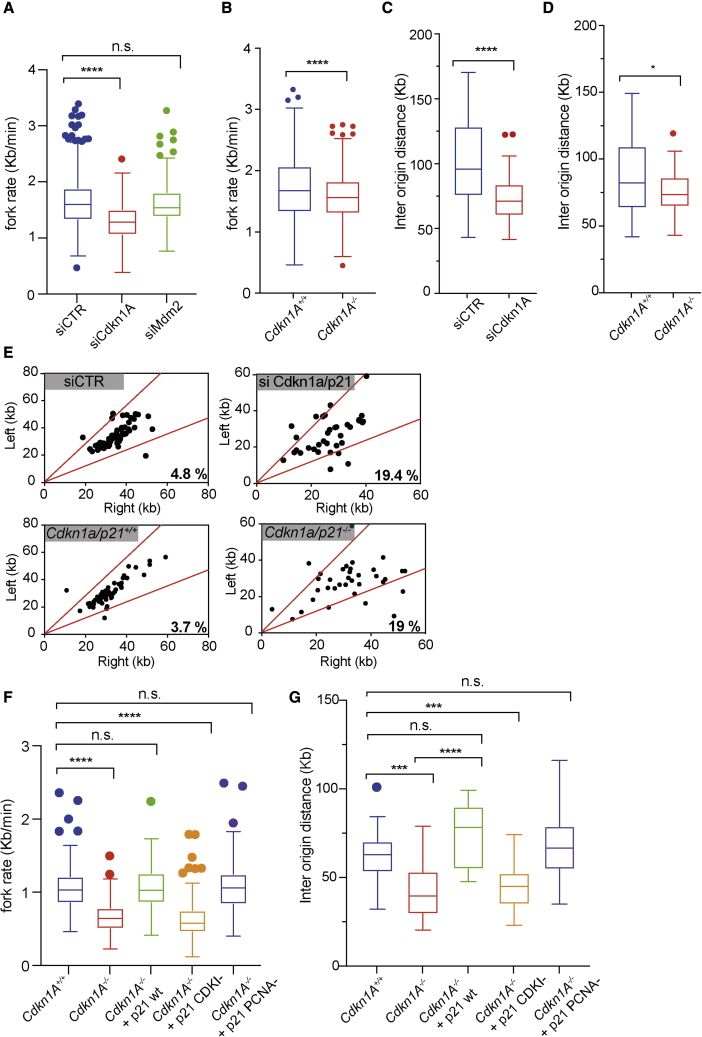
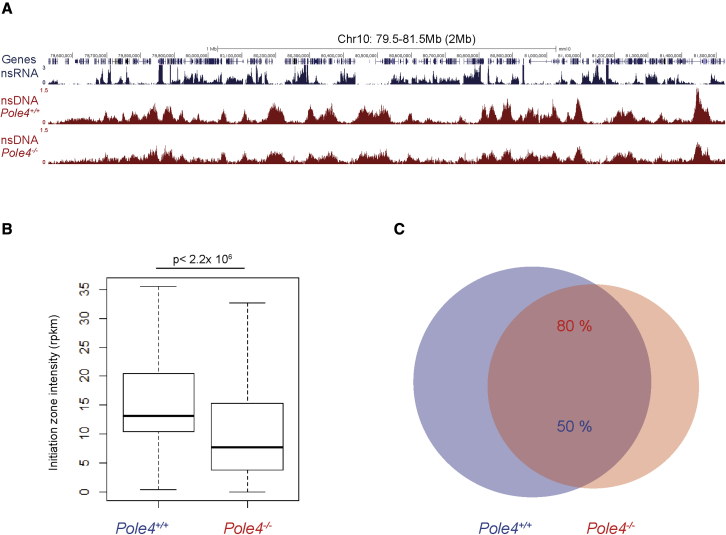
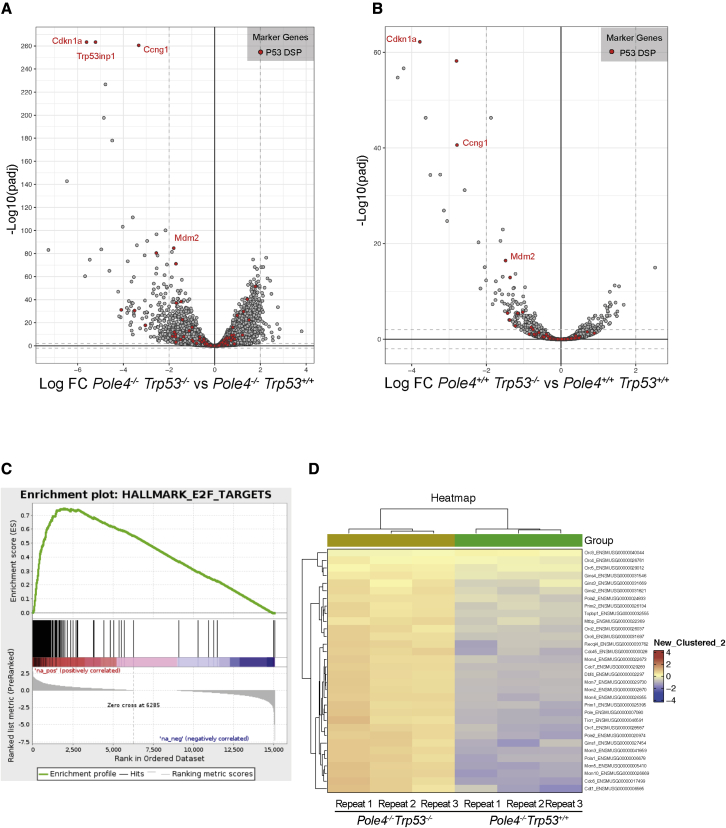
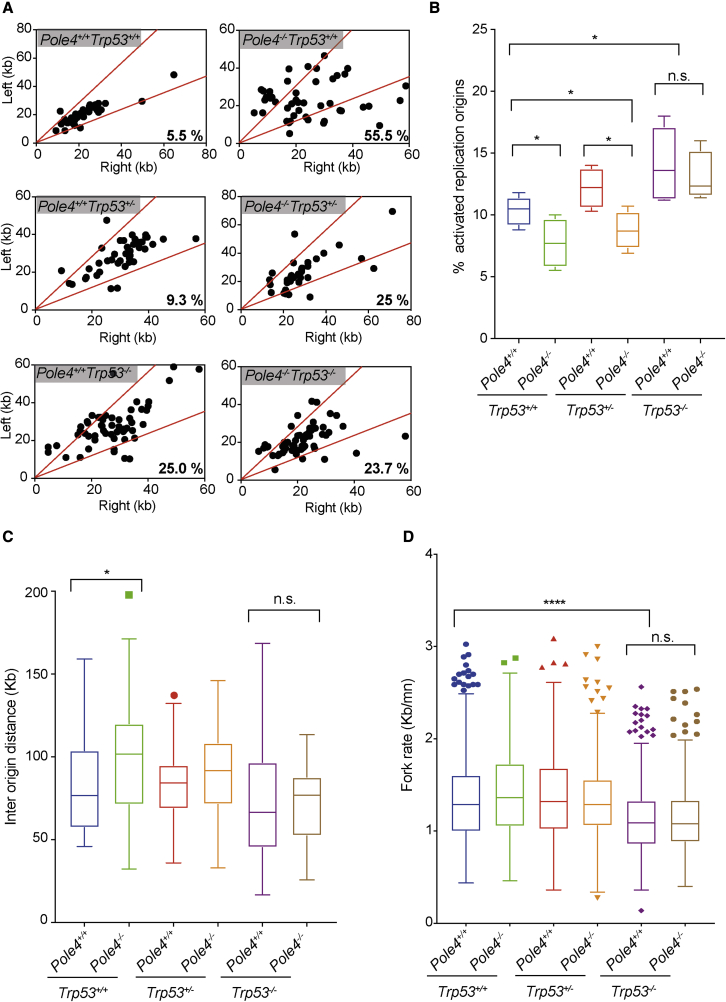
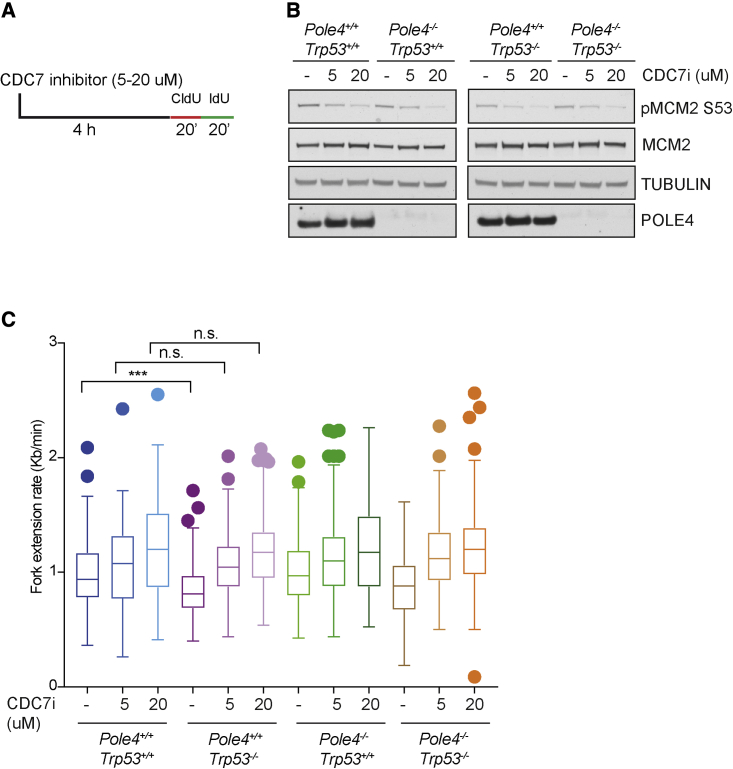
The maintenance of genome stability relies on coordinated control of origin activation and replication fork progression. How the interplay between these processes influences human genetic disease and cancer remains incompletely characterized. Here we show that mouse cells featuring Polε instability exhibit impaired genome-wide activation of DNA replication origins, in an origin-location-independent manner. Strikingly, Trp53 ablation in primary Polε hypomorphic cells increased Polε levels and origin activation and reduced DNA damage in a transcription-dependent manner. Transcriptome analysis of primary Trp53 knockout cells revealed that the TRP53-CDKN1A/P21 axis maintains appropriate levels of replication factors and CDK activity during unchallenged S phase. Loss of this control mechanism deregulates origin activation and perturbs genome-wide replication fork progression. Thus, while our data support an impaired origin activation model for genetic diseases affecting CMG formation, we propose that loss of the TRP53-CDKN1A/P21 tumor suppressor axis induces inappropriate origin activation and deregulates genome-wide fork progression.
基因组稳定性的维持依赖于对起始点激活和复制叉进展的协调控制。这些过程之间的相互作用如何影响人类遗传疾病和癌症,目前仍不完全清楚。在这里,我们表明,具有Polε不稳定性的小鼠细胞以一种不依赖于起始点位置的方式,在全基因组范围内的DNA复制起始点激活方面存在缺陷。令人惊讶的是,原代Polε低表达细胞中的Trp53缺失以转录依赖的方式增加了Polε水平和起始点激活,并减少了DNA损伤。对原代Trp53基因敲除细胞的转录组分析表明,TRP53-CDKN1A/P21轴在未受挑战的S期维持复制因子和CDK活性的适当水平。这种控制机制的丧失会使起始点激活失调,并扰乱全基因组范围内的复制叉进展。因此,虽然我们的数据支持一种影响CMG形成的遗传疾病的起始点激活受损模型,但我们提出,TRP53-CDKN1A/P21肿瘤抑制轴的丧失会导致不适当的起始点激活,并使全基因组范围内的复制叉进展失调。