Division of Molecular Medicine, Leeds Institute of Medical Research, St. James's University Hospital, University of Leeds, Leeds, UK.
Department of Zoology, Faculty of Science, Benha University, Benha, Egypt.
Mol Vis. 2022 May 17;28:48-56. eCollection 2022.
To describe the clinical phenotype and genetic basis of non-syndromic retinitis pigmentosa (RP) in one family and two sporadic cases with biallelic mutations in the transcription factor neural retina leucine zipper (.
Exome sequencing was performed in one affected family member. Microsatellite genotyping was used for haplotype analysis. PCR and Sanger sequencing were used to confirm mutations in and screen other family members where they were available. The SMART tool for domain prediction helped us build the protein schematic diagram.
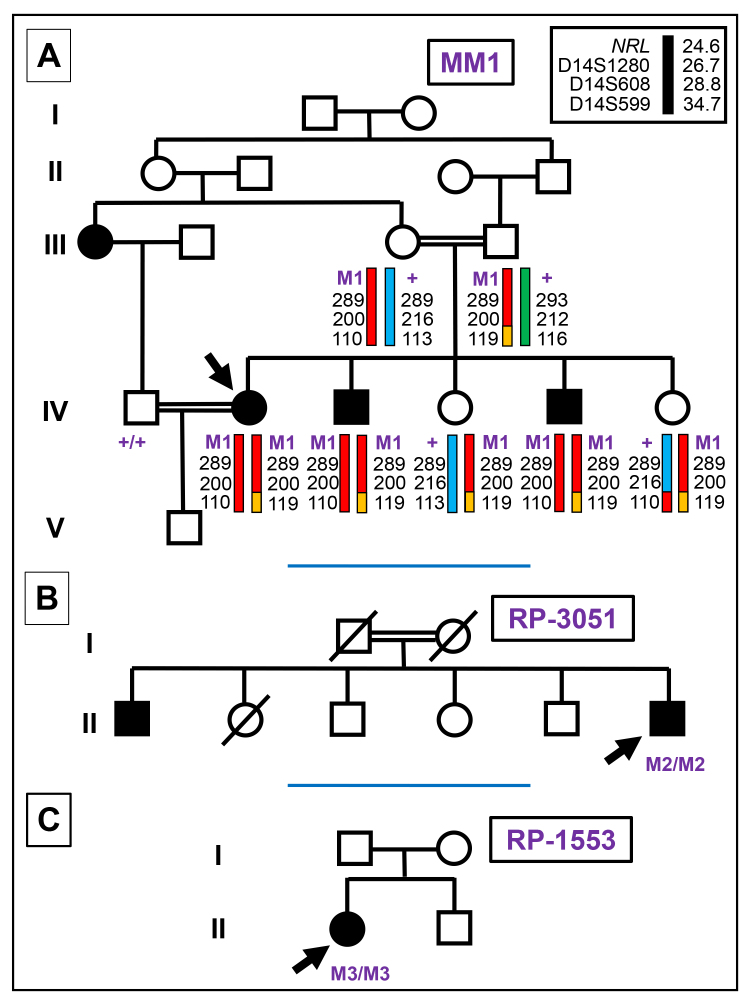
For family MM1 of Pakistani origin, whole-exome sequencing and microsatellite genotyping revealed homozygosity on chromosome 14 and identified a homozygous stop-loss mutation in , NM_006177.5: c.713G>T, p.238Lext57, which is predicted to add an extra 57 amino acids to the normal protein chain. The variant segregated with disease symptoms in the family. For case RP-3051 of Spanish ancestry, clinical exome sequencing focusing on the morbid genome highlighted a homozygous nonsense mutation in , c.238C>T, p.Gln80, as the most likely disease candidate. For case RP-1553 of Romanian ethnicity, targeted-exome sequencing of 73 RP/LCA genes identified a homozygous nonsense mutation in , c.544C>T, p.Gln182*. The variants were either rare or absent in the gnomAD database.
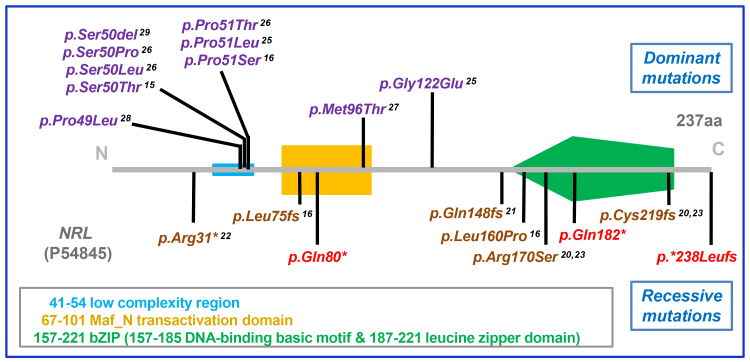
mutations predominantly cause dominant retinal disease, but there have been five published reports of mutations causing recessive disease. Here, we present three further examples of recessive RP due to mutations. The phenotypes observed are consistent with those in the previous reports, and the observed mutation types and distribution further confirm distinct patterns for variants in causing recessive and dominant diseases.
描述一个家系和两个散发病例中非综合征性视网膜色素变性(RP)的临床表型和遗传基础,这些病例在转录因子神经视网膜亮氨酸拉链( 中存在双等位基因突变。
对一个受影响的家系成员进行外显子组测序。微卫星基因分型用于单倍型分析。PCR 和 Sanger 测序用于确认和筛选其他有条件的家系成员中的突变。用于结构域预测的 SMART 工具帮助我们构建了蛋白质示意图。
对于来自巴基斯坦的 MM1 家系,全外显子组测序和微卫星基因分型显示 14 号染色体纯合子,并在 中发现一个纯合无义突变,NM_006177.5:c.713G>T,p.238Lext57,预计会在正常蛋白链上添加 57 个额外的氨基酸。该变体在家系中与疾病症状共分离。对于来自西班牙的 RP-3051 病例,针对病态基因组的临床外显子组测序突出显示 中的一个纯合无义突变,c.238C>T,p.Gln80,是最有可能的疾病候选者。对于罗马尼亚血统的 RP-1553 病例,73 个 RP/LCA 基因的靶向外显子组测序鉴定出 中的一个纯合无义突变,c.544C>T,p.Gln182*。变体在 gnomAD 数据库中要么是罕见的,要么是不存在的。
突变主要导致显性视网膜疾病,但已有五份关于突变导致隐性疾病的报道。在这里,我们提出了三个由于 突变导致隐性 RP 的进一步例子。观察到的表型与之前的报告一致,观察到的突变类型和分布进一步证实了 导致隐性和显性疾病的变体的不同模式。