Evolutionary Genomics Group, División de Microbiología, Universidad Miguel Hernández, San Juan, Alicante, Spain.
mSystems. 2022 Jun 28;7(3):e0019222. doi: 10.1128/msystems.00192-22. Epub 2022 Jun 13.
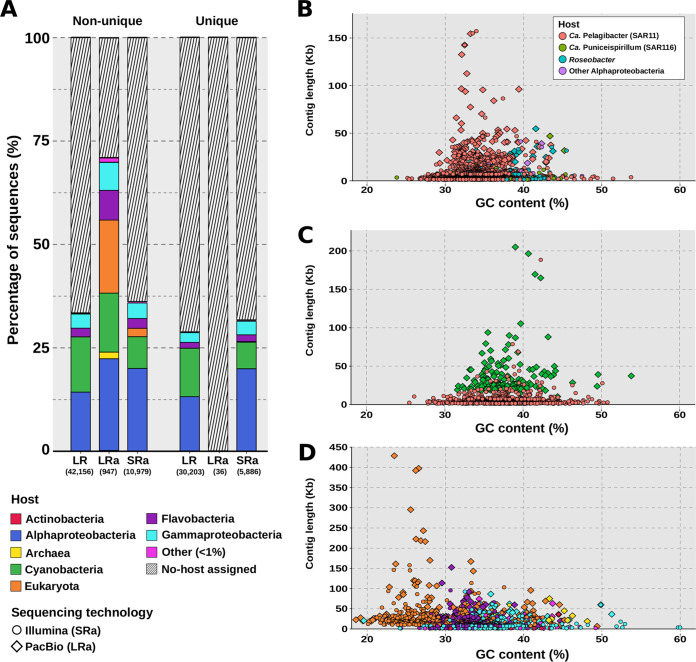
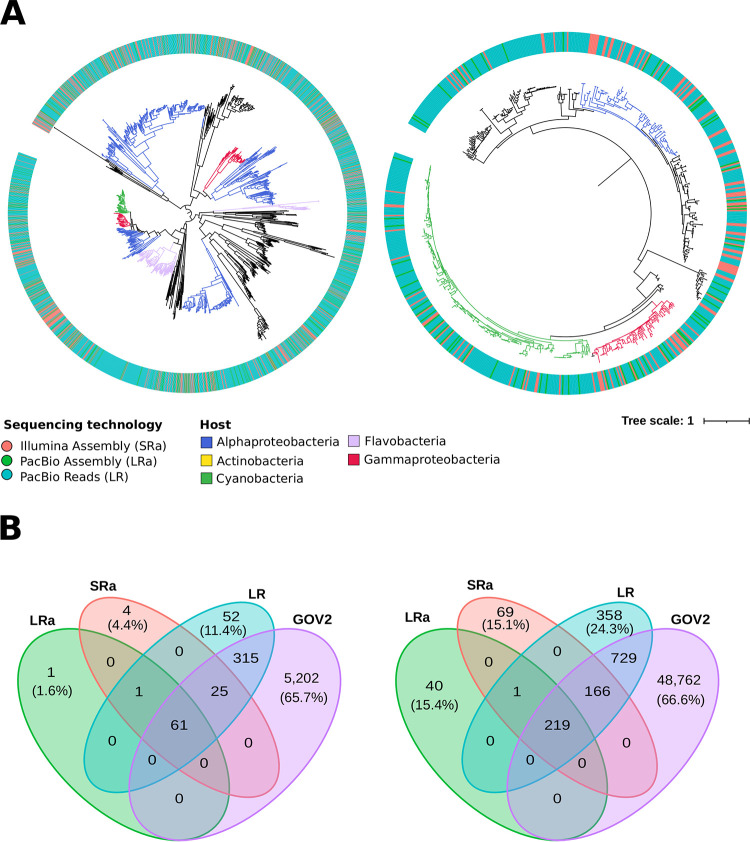
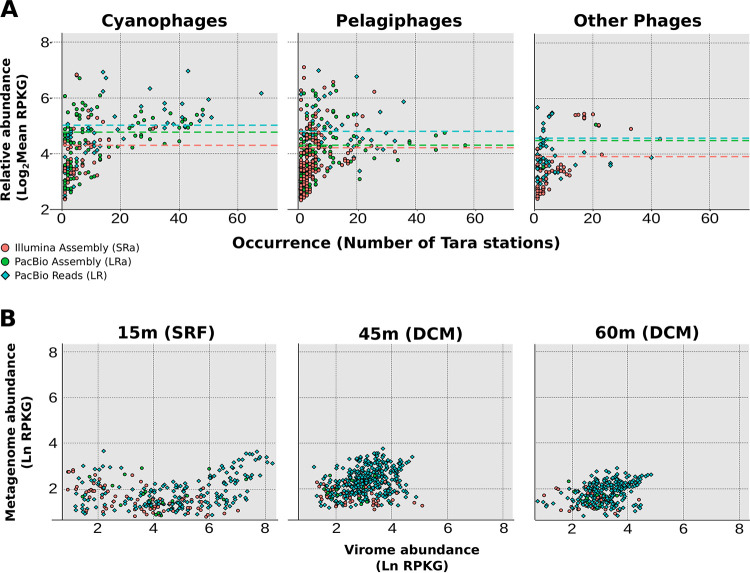
The recovery of DNA from viromes is a major obstacle in the use of long-read sequencing to study their genomes. For this reason, the use of cellular metagenomes (>0.2-μm size range) emerges as an interesting complementary tool, since they contain large amounts of naturally amplified viral genomes from prelytic replication. We have applied second-generation (Illumina NextSeq; short reads) and third-generation (PacBio Sequel II; long reads) sequencing to compare the diversity and features of the viral community in a marine sample obtained from offshore waters of the western Mediterranean. We found that a major wedge of the expected marine viral diversity was directly recovered by the raw PacBio circular consensus sequencing (CCS) reads. More than 30,000 sequences were detected only in this data set, with no homologues in the long- and short-read assembly, and ca. 26,000 had no homologues in the large data set of the Global Ocean Virome 2 (GOV2), highlighting the information gap created by the assembly bias. At the level of complete viral genomes, the performance was similar in both approaches. However, the hybrid long- and short-read assembly provided the longest average length of the sequences and improved the host assignment. Although no novel major clades of viruses were found, there was an increase in the intraclade genomic diversity recovered by long reads that produced an enriched assessment of the real diversity and allowed the discovery of novel genes with biotechnological potential (e.g., endolysin genes). We explored the vast genetic diversity of environmental viruses by using a combination of cellular metagenome (as opposed to virome) sequencing using high-fidelity long-read sequences (in this case, PacBio CCS). This approach resulted in the recovery of a representative sample of the viral population, and it performed better (more phage contigs, larger average contig size) than Illumina sequencing applied to the same sample. By this approach, the many biases of assembly are avoided, as the CCS reads recovers (typically around 5 kb) complete genes and even operons, resulting in a better discovery of the viral gene diversity based on viral marker proteins. Thus, biotechnologically promising genes, such as endolysin genes, can be very efficiently searched with this approach. In addition, hybrid assembly produces more complete and longer contigs, which is particularly important for studying little-known viral groups such as the nucleocytoplasmic large DNA viruses (NCLDV).
从病毒组中回收 DNA 是使用长读测序研究其基因组的主要障碍。出于这个原因,使用细胞宏基因组(>0.2-μm 大小范围)作为一种有趣的互补工具出现了,因为它们包含大量来自预溶复制的天然扩增病毒基因组。我们应用第二代(Illumina NextSeq;短读长)和第三代(PacBio Sequel II;长读长)测序技术,比较了从地中海西部近海海域获得的海洋样本中病毒群落的多样性和特征。我们发现,通过原始的 PacBio 圆形一致测序(CCS)reads 可以直接回收预期海洋病毒多样性的主要部分。在这个数据集仅检测到超过 30000 个序列,在长读和短读组装中没有同源物,大约 26000 个序列在大型全球海洋病毒组 2(GOV2)数据集中没有同源物,突出了组装偏差造成的信息缺口。在完整病毒基因组水平上,两种方法的性能相似。然而,长读和短读混合组装提供了最长的序列平均长度,并改善了宿主分配。虽然没有发现新的主要病毒进化枝,但长读产生的进化枝内基因组多样性增加,从而对真实多样性进行了更丰富的评估,并发现了具有生物技术潜力的新基因(例如,内溶素基因)。我们通过使用高保真度长读序列(在这种情况下为 PacBio CCS)对细胞宏基因组(而不是病毒组)进行测序,探索了环境病毒的巨大遗传多样性。这种方法导致了病毒群体的代表性样本的回收,并且它比应用于相同样本的 Illumina 测序表现更好(更多噬菌体 contigs,更大的平均 contig 大小)。通过这种方法,可以避免许多组装偏差,因为 CCS reads 回收(通常约 5kb)完整的基因甚至操纵子,从而基于病毒标记蛋白更好地发现病毒基因多样性。因此,可以非常有效地使用这种方法搜索具有生物技术前景的基因,例如内溶素基因。此外,混合组装产生更完整和更长的 contigs,这对于研究核质大 DNA 病毒(NCLDV)等鲜为人知的病毒群特别重要。