Muntean Carmen, Starcea Iuliana Magdalena, Stoica Cristina, Banescu Claudia
Department of Pediatrics I, George Emil Palade University of Medicine, Pharmacy, Science, and Technology of Targu Mures, Targu Mures, Romania.
Department of Pediatric Nephrology, Sf Maria Emergency Hospital for Children Iasi, University of Medicine and Pharmacy Grigore T. Popa Iasi, Iasi, Romania.
Front Pediatr. 2022 Jun 1;10:908657. doi: 10.3389/fped.2022.908657. eCollection 2022.
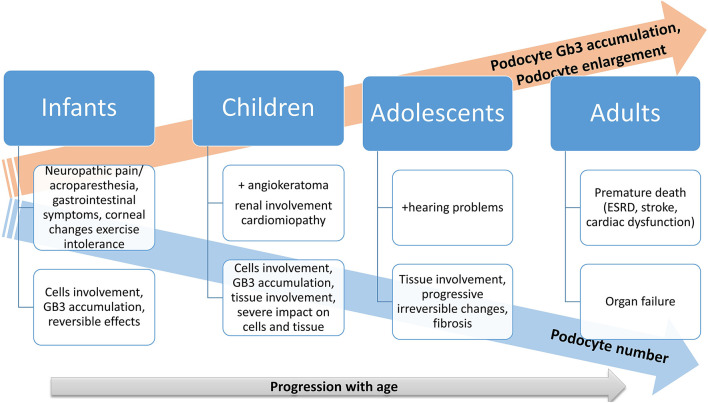
Inherited renal diseases represent 20% of the causes of end-stage renal diseases. Fabry disease, an X-linked lysosomal storage disorder, results from α-galactosidase A deficient or absent activity followed by globotriaosylceramide (Gb3) accumulation and multiorgan involvement. In Fabry disease, kidney involvement starts early, during intrauterine life by the Gb3 deposition. Even if chronic kidney disease (CKD) is discovered later in adult life in Fabry disease patients, a decline in glomerular filtration rate (GFR) can occur during adolescence. The first clinical sign of kidney involvement is represented by albuminuria. So, early and close monitoring of kidneys function is required: albuminuria and proteinuria, urinary albumin-to-creatinine ratio, serum creatinine, or cystatin C to estimate GFR, while urinary sediment with phase-contrast microscopy under polarized light may be useful in those cases where leucocyte α-Gal A activity and genotyping are not available. Children with Fabry disease and kidney involvement should receive enzyme replacement therapy and nephroprotective drugs (angiotensin-converting enzyme inhibitors or angiotensin receptor blockers) to prevent or slow the progressive loss of kidney functions. Early diagnosis of Fabry disease is important as enzyme replacement therapy reduces symptoms, improves clinical features and biochemical markers, and the quality of life. More importantly, early treatment could slow or stop progressive organ damage in later life.
遗传性肾病占终末期肾病病因的20%。法布里病是一种X连锁溶酶体贮积症,由α-半乳糖苷酶A活性缺乏或缺失导致球三糖神经酰胺(Gb3)蓄积及多器官受累。在法布里病中,肾脏受累始于子宫内生活期间Gb3的沉积。即使法布里病患者在成年后才发现慢性肾脏病(CKD),在青春期肾小球滤过率(GFR)也可能下降。肾脏受累的首个临床症状为蛋白尿。因此,需要对肾功能进行早期密切监测:监测蛋白尿和蛋白尿、尿白蛋白与肌酐比值、血清肌酐或胱抑素C以评估GFR,而在无法检测白细胞α-Gal A活性和基因分型的情况下,偏振光下相差显微镜检查尿沉渣可能有用。患有法布里病且肾脏受累的儿童应接受酶替代疗法和肾脏保护药物(血管紧张素转换酶抑制剂或血管紧张素受体阻滞剂),以预防或减缓肾功能的进行性丧失。法布里病的早期诊断很重要,因为酶替代疗法可减轻症状、改善临床特征和生化指标以及提高生活质量。更重要的是,早期治疗可减缓或阻止后期生活中器官的进行性损害。