Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA, USA.
Harvard-MIT Health Sciences and Technology Program, Cambridge, MA, USA.
Nat Biotechnol. 2022 Nov;40(11):1634-1643. doi: 10.1038/s41587-022-01353-8. Epub 2022 Jun 20.
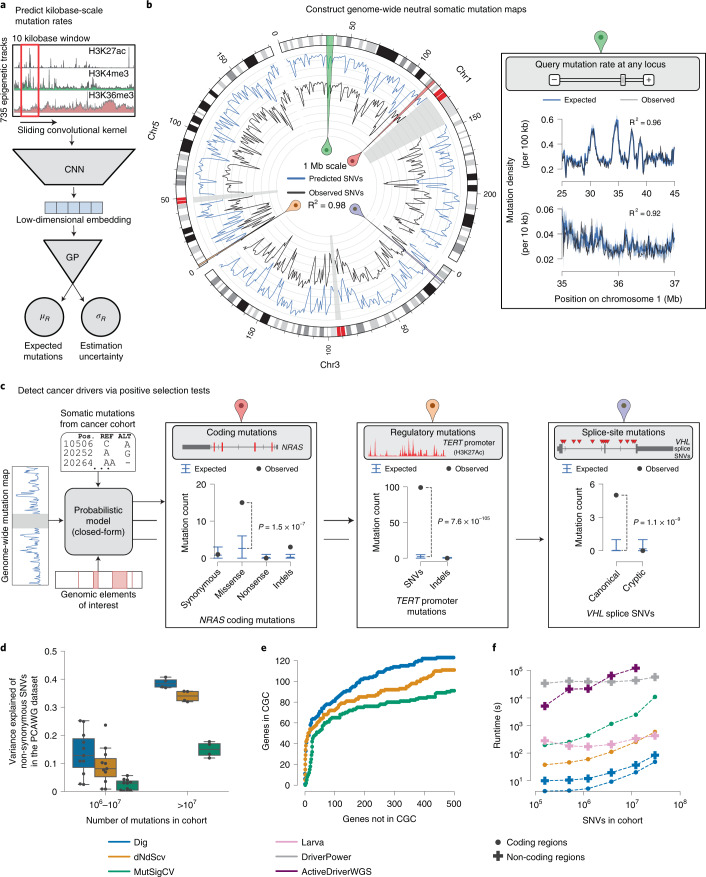
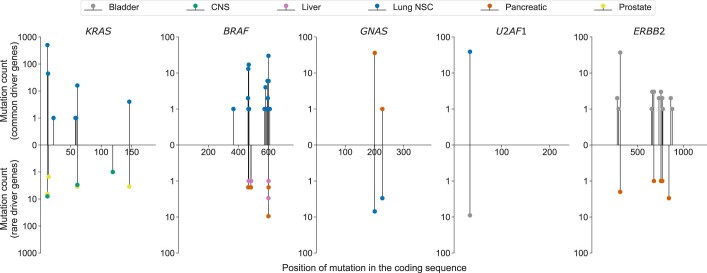
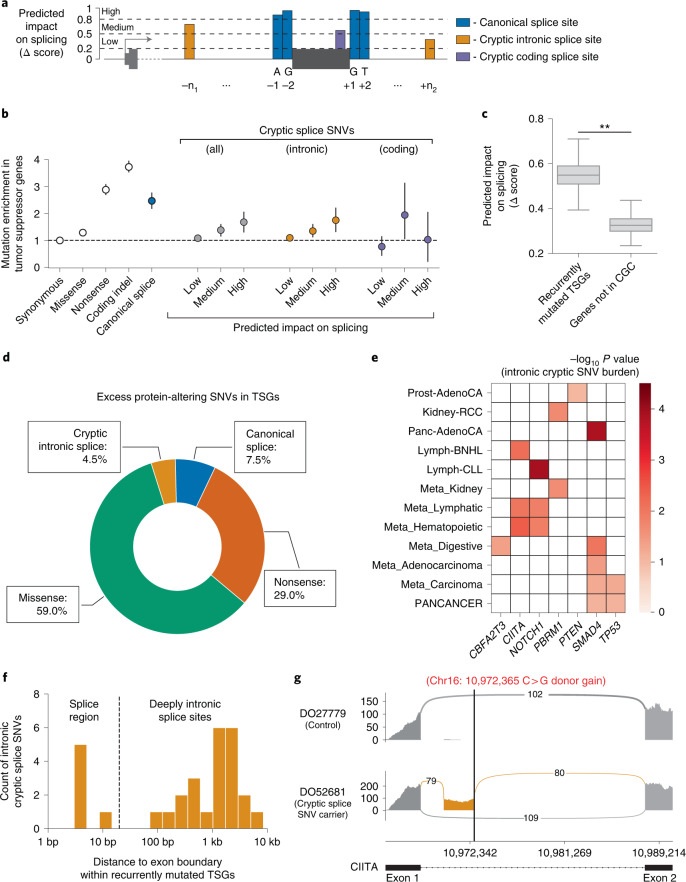
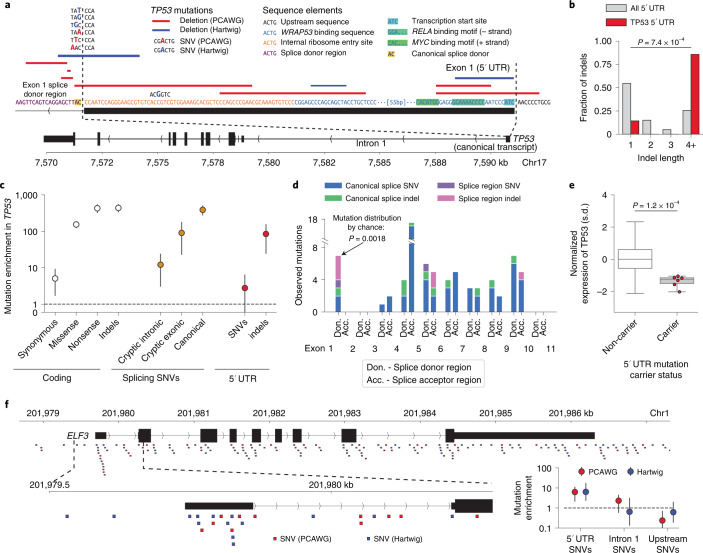
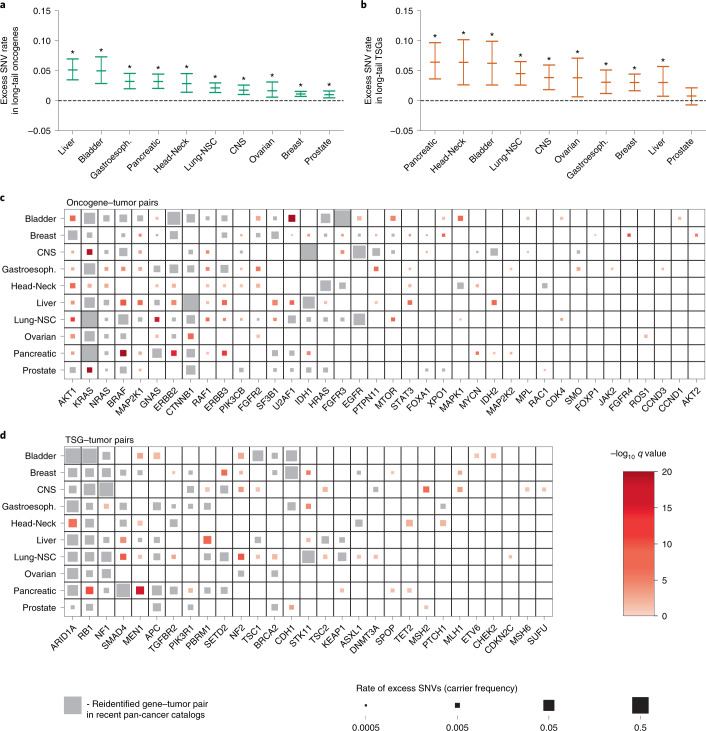
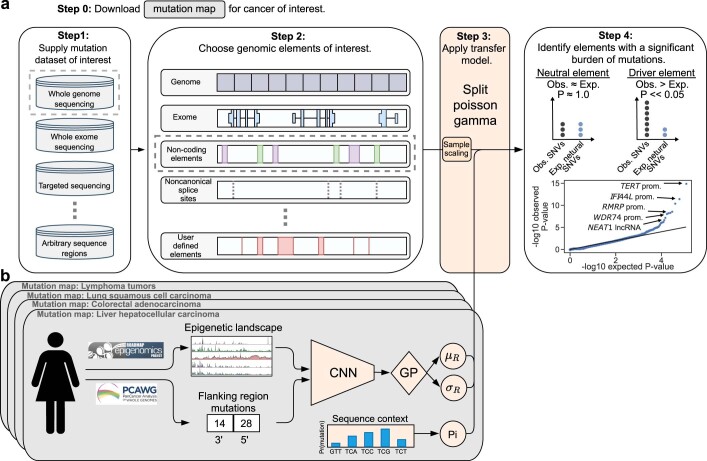
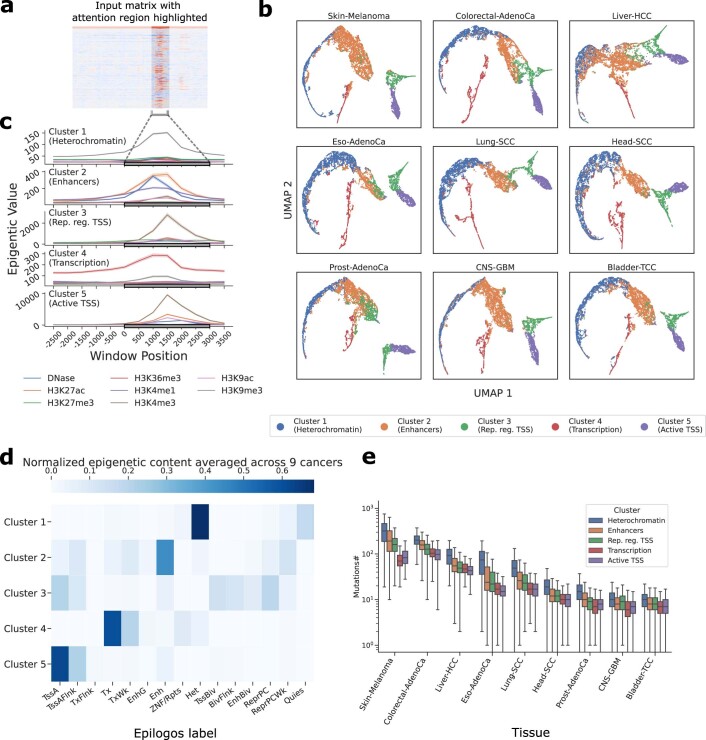
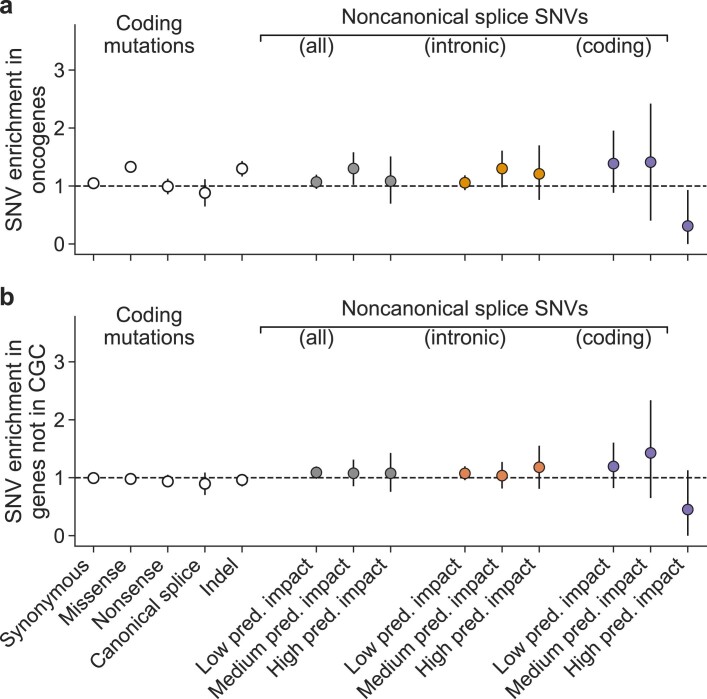
Identification of cancer driver mutations that confer a proliferative advantage is central to understanding cancer; however, searches have often been limited to protein-coding sequences and specific non-coding elements (for example, promoters) because of the challenge of modeling the highly variable somatic mutation rates observed across tumor genomes. Here we present Dig, a method to search for driver elements and mutations anywhere in the genome. We use deep neural networks to map cancer-specific mutation rates genome-wide at kilobase-scale resolution. These estimates are then refined to search for evidence of driver mutations under positive selection throughout the genome by comparing observed to expected mutation counts. We mapped mutation rates for 37 cancer types and applied these maps to identify putative drivers within intronic cryptic splice regions, 5' untranslated regions and infrequently mutated genes. Our high-resolution mutation rate maps, available for web-based exploration, are a resource to enable driver discovery genome-wide.
鉴定赋予增殖优势的癌症驱动突变是理解癌症的核心;然而,由于难以对肿瘤基因组中观察到的高度可变的体细胞突变率进行建模,搜索通常仅限于蛋白质编码序列和特定的非编码元件(例如启动子)。在这里,我们提出了 Dig,这是一种在基因组的任何位置搜索驱动元件和突变的方法。我们使用深度神经网络在千碱基分辨率的全基因组范围内绘制癌症特异性突变率图。然后,通过将观察到的突变计数与预期的突变计数进行比较,对这些估计值进行细化,以在整个基因组中搜索正选择驱动突变的证据。我们绘制了 37 种癌症类型的突变率图,并将这些图谱应用于识别内含子隐匿剪接区、5'非翻译区和突变频率较低的基因中的潜在驱动因子。我们的高分辨率突变率图谱可在网上进行探索,是一个有助于在全基因组范围内发现驱动因子的资源。