Khade Pranav M, Jernigan Robert L
Bioinformatics and Computational Biology Program and Roy J. Carver Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, Iowa 50011, United States.
ACS Omega. 2022 Jun 9;7(24):20719-20730. doi: 10.1021/acsomega.2c00999. eCollection 2022 Jun 21.
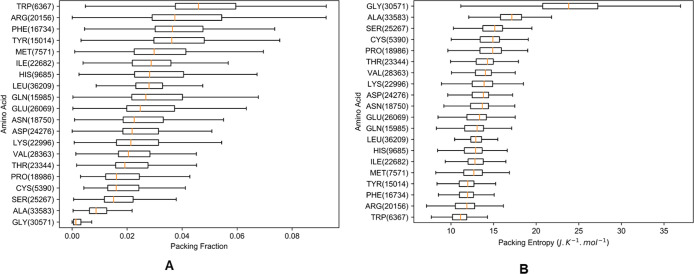
A fast, simple, yet robust method to calculate protein entropy from a single protein structure is presented here. The focus is on the atomic packing details, which are calculated by combining Voronoi diagrams and Delaunay tessellations. Even though the method is simple, the entropies computed exhibit an extremely high correlation with the entropies previously derived by other methods based on quasi-harmonic motions, quantum mechanics, and molecular dynamics simulations. These packing-based entropies account directly for the local freedom and provide entropy for any individual protein structure that could be used to compute free energies directly during simulations for the generation of more reliable trajectories and also for better evaluations of modeled protein structures. Physico-chemical properties of amino acids are compared with these packing entropies to uncover the relationships with the entropies of different residue types. A public packing entropy web server is provided at packing-entropy.bb.iastate.edu, and the application programing interface is available within the PACKMAN (https://github.com/Pranavkhade/PACKMAN) package.
本文介绍了一种从单个蛋白质结构计算蛋白质熵的快速、简单且稳健的方法。重点在于原子堆积细节,通过结合Voronoi图和Delaunay三角剖分来计算。尽管该方法简单,但计算出的熵与先前通过基于准谐运动、量子力学和分子动力学模拟的其他方法得出的熵具有极高的相关性。这些基于堆积的熵直接考虑了局部自由度,并为任何单个蛋白质结构提供熵,可用于在模拟过程中直接计算自由能,以生成更可靠的轨迹,还可用于更好地评估建模的蛋白质结构。将氨基酸的物理化学性质与这些堆积熵进行比较,以揭示与不同残基类型熵的关系。在packing - entropy.bb.iastate.edu提供了一个公共的堆积熵网络服务器,并且在PACKMAN(https://github.com/Pranavkhade/PACKMAN)软件包中提供了应用程序编程接口。