Department of Computer Science and Engineering, The Pennsylvania State University, University Park, PA, USA.
Department of Biology, The Pennsylvania State University, University Park, PA, USA.
Bioinformatics. 2022 Jun 24;38(Suppl 1):i169-i176. doi: 10.1093/bioinformatics/btac244.
Sketching is now widely used in bioinformatics to reduce data size and increase data processing speed. Sketching approaches entice with improved scalability but also carry the danger of decreased accuracy and added bias. In this article, we investigate the minimizer sketch and its use to estimate the Jaccard similarity between two sequences.
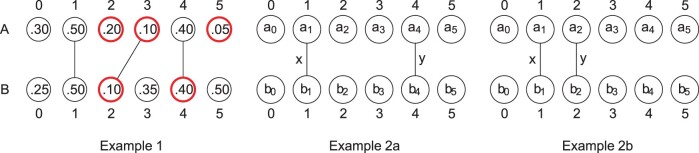
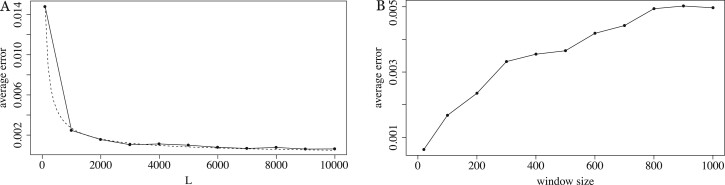
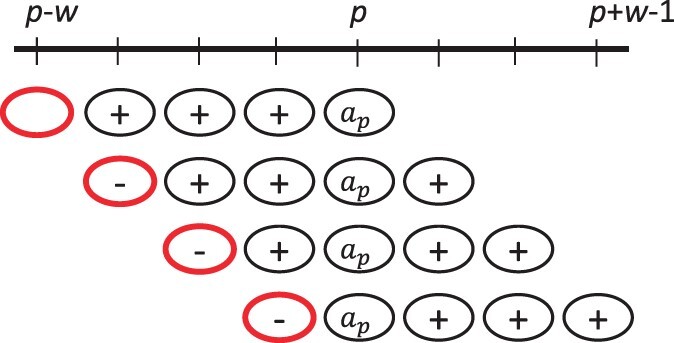
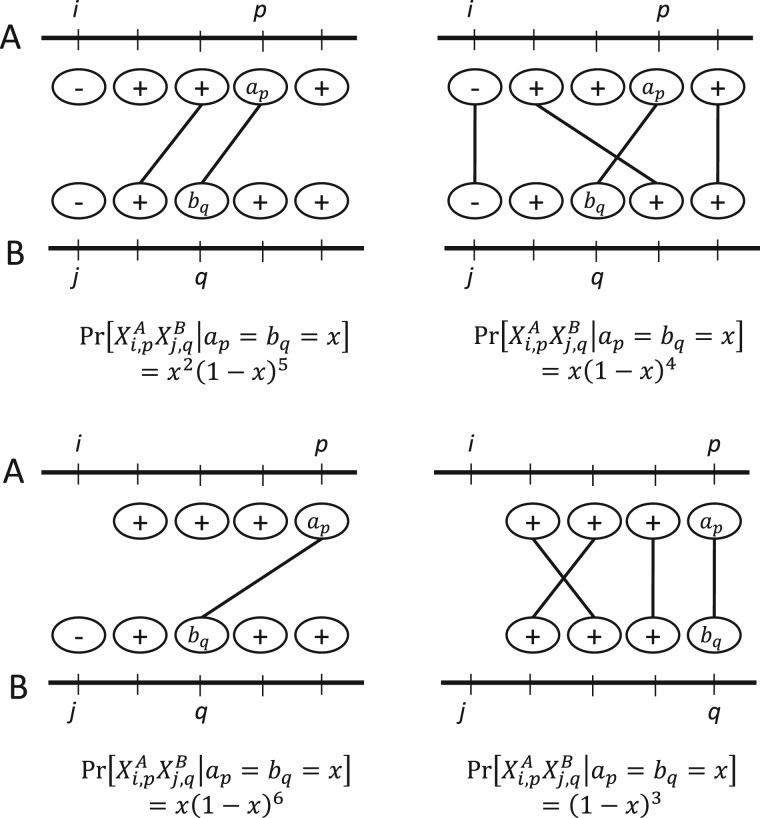
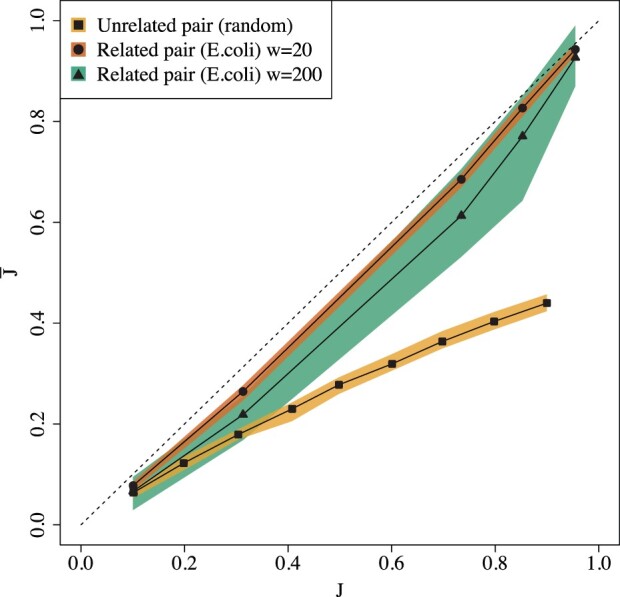
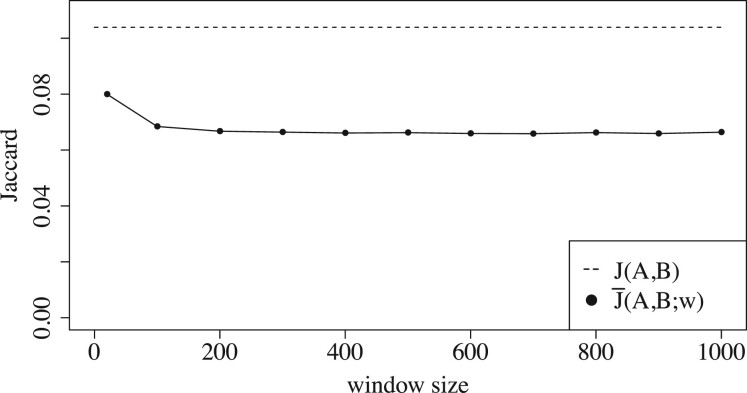
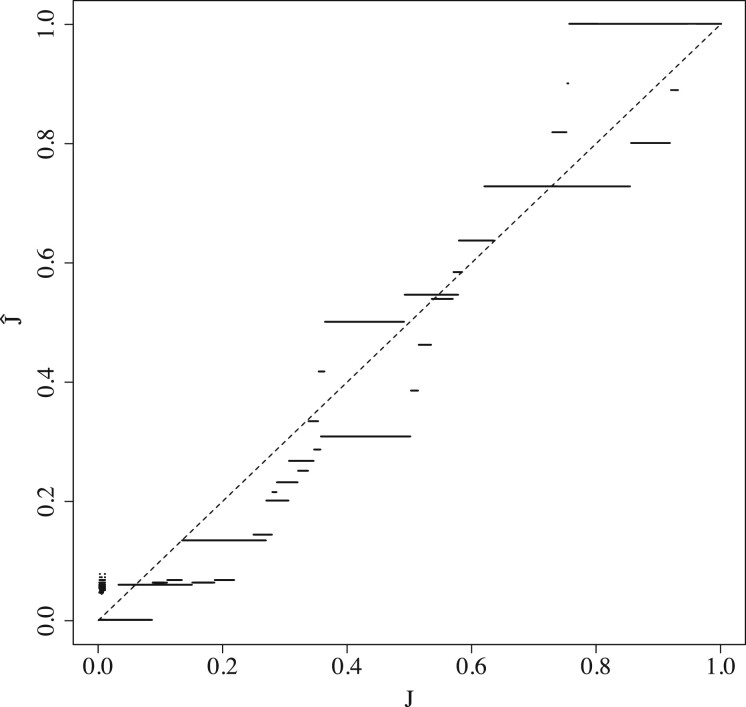
We show that the minimizer Jaccard estimator is biased and inconsistent, which means that the expected difference (i.e. the bias) between the estimator and the true value is not zero, even in the limit as the lengths of the sequences grow. We derive an analytical formula for the bias as a function of how the shared k-mers are laid out along the sequences. We show both theoretically and empirically that there are families of sequences where the bias can be substantial (e.g. the true Jaccard can be more than double the estimate). Finally, we demonstrate that this bias affects the accuracy of the widely used mashmap read mapping tool.
Scripts to reproduce our experiments are available at https://github.com/medvedevgroup/minimizer-jaccard-estimator/tree/main/reproduce.
Supplementary data are available at Bioinformatics online.
现在草图在生物信息学中被广泛用于减少数据量和提高数据处理速度。草图方法具有改进的可扩展性,但也存在准确性降低和偏差增加的风险。在本文中,我们研究了最小器草图及其在估计两个序列之间的 Jaccard 相似性中的应用。
我们表明最小器 Jaccard 估计器存在偏差和不一致性,这意味着估计器与真实值之间的期望差异(即偏差)不为零,即使在序列长度增长的极限情况下也是如此。我们推导出了一个偏差的解析公式,作为共享 k-mer 沿着序列排列方式的函数。我们从理论和实验上都表明,存在一些序列族,其中偏差可能很大(例如,真实的 Jaccard 可以是估计值的两倍以上)。最后,我们证明了这种偏差会影响广泛使用的 mashmap 读取映射工具的准确性。
可在 https://github.com/medvedevgroup/minimizer-jaccard-estimator/tree/main/reproduce 获得重现我们实验的脚本。
补充数据可在 Bioinformatics 在线获得。