Immunology Division, Vall d'Hebron University Hospital, Universitat Autònoma de Barcelona (UAB), Barcelona, Spain.
Translational Immunology Research Group, Vall d'Hebron Research Institute (VHIR), Vall d'Hebron University Hospital, Barcelona, Spain.
Front Immunol. 2022 Jun 17;13:897975. doi: 10.3389/fimmu.2022.897975. eCollection 2022.
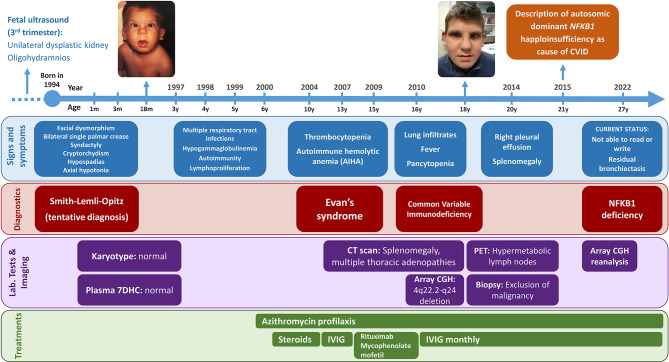
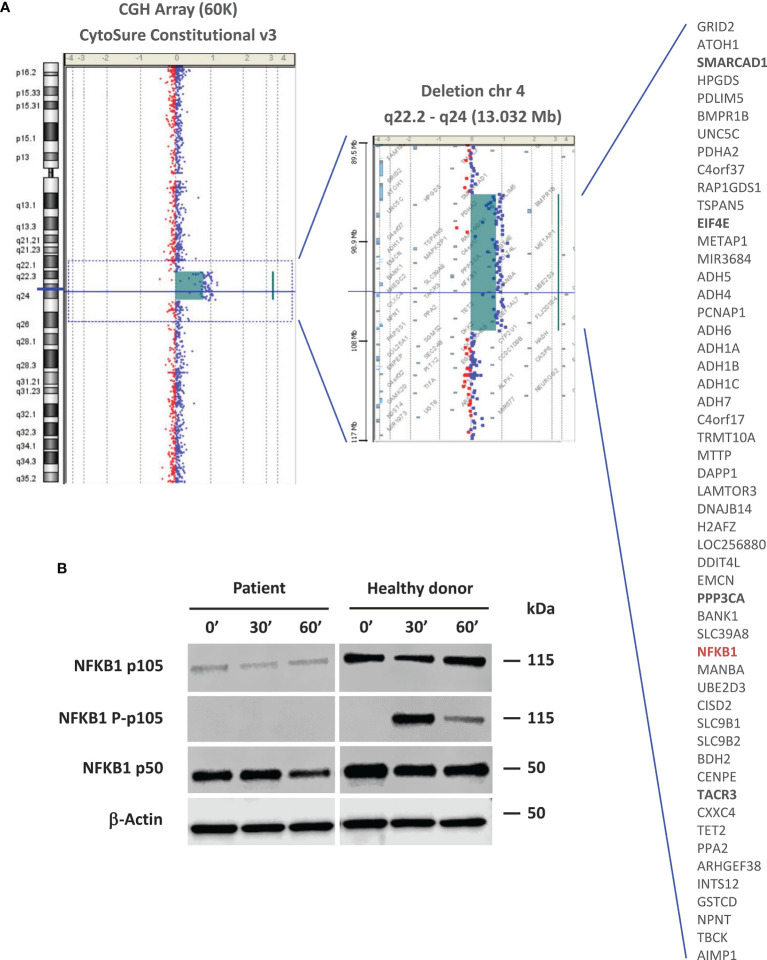
Syndromic immunodeficiencies are a heterogeneous group of inborn errors of immunity that can affect the development of non-immune organs and systems. The genetic basis of these immunodeficiencies is highly diverse, ranging from monogenic defects to large chromosomal aberrations. Antibody deficiency is the most prevalent immunological abnormality in patients with syndromic immunodeficiencies caused by chromosomal rearrangements, and usually manifests as a common variable immunodeficiency (CVID)-like phenotype. Here we describe a patient with a complex phenotype, including neurodevelopmental delay, dysmorphic features, malformations, and CVID (hypogammaglobulinemia, reduced pre-switch and switch memory B cells, and impaired vaccine response). Microarray-based comparative genomic hybridization (aCGH) revealed a 13-Mb deletion on chromosome 4q22.2-q24 involving 53 genes, some of which were related to the developmental manifestations in our patient. Although initially none of the affected genes could be linked to his CVID phenotype, subsequent reanalysis identified haploinsufficiency as the cause. This study underscores the value of periodic reanalysis of unsolved genetic studies performed with high-throughput technologies (eg, next-generation sequencing and aCGH). This is important because of the ongoing incorporation of new data establishing the relationship between genes and diseases. In the present case, had not been associated with human disease at the time aCGH was performed. Eight years later, reanalysis of the genes included in the chromosome 4 deletion enabled us to identify haploinsufficiency as the genetic cause of our patient's CVID. In the future, other genes included in the deletion may be linked to human disease, allowing us to better define the molecular basis of our patient's complex clinical phenotype.
综合征性免疫缺陷是一组异质性的先天性免疫缺陷,可影响非免疫器官和系统的发育。这些免疫缺陷的遗传基础高度多样化,从单基因缺陷到大片段染色体异常都有涉及。抗体缺陷是由染色体重排引起的综合征性免疫缺陷患者中最常见的免疫异常,通常表现为普通变异型免疫缺陷(CVID)样表型。在这里,我们描述了一例具有复杂表型的患者,包括神经发育迟缓、畸形特征、畸形和 CVID(低丙种球蛋白血症、减少前转换和转换记忆 B 细胞以及疫苗反应受损)。基于微阵列的比较基因组杂交(aCGH)显示,4q22.2-q24 染色体上有 13Mb 的缺失,涉及 53 个基因,其中一些与我们患者的发育表现有关。虽然最初没有一个受影响的基因与他的 CVID 表型有关,但随后的重新分析确定了杂合性不足是原因。本研究强调了定期重新分析高通量技术(如下一代测序和 aCGH)进行的未解决遗传研究的价值。这很重要,因为新数据不断确立基因与疾病之间的关系。在本例中,在进行 aCGH 时, 尚未与人类疾病相关联。八年后,对包括在染色体 4 缺失中的基因进行重新分析,使我们能够确定 杂合性不足是我们患者 CVID 的遗传原因。在未来,缺失中包含的其他基因可能与人类疾病有关,使我们能够更好地定义我们患者复杂临床表型的分子基础。