Tsai Erin, Gallage Dona Hishara Keshani, Tong Xinjie, Du Pu, Novak Brian, David Rolf, Rick Steven W, Zhang Donghui, Kumar Revati
Department of Chemistry, Louisiana State University, Baton Rouge, Louisiana 70803, United States.
Department of Mechanical and Industrial Engineering, Louisiana State University, Baton Rouge, Louisiana 70803, United States.
Macromolecules. 2022 Jun 28;55(12):5197-5212. doi: 10.1021/acs.macromol.2c00141. Epub 2022 Jun 14.
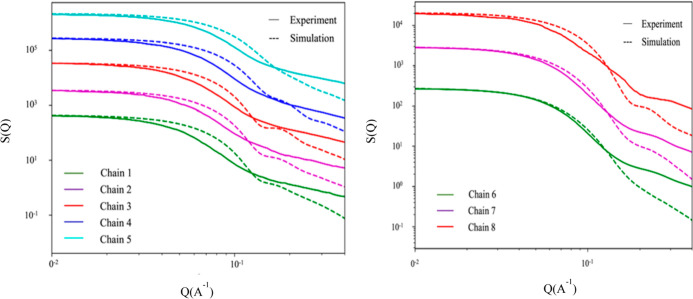
Electrostatic interactions play a significant role in regulating biological systems and have received increasing attention due to their usefulness in designing advanced stimulus-responsive materials. Polypeptoids are highly tunable N-substituted peptidomimetic polymers that lack backbone hydrogen bonding and chirality. Therefore, polypeptoids are suitable systems to study the effect of noncovalent interactions of substituents without complications of backbone intramolecular and intermolecular hydrogen bonding. In this study, all-atom molecular dynamics (MD) simulations were performed on micelles formed by a series of sequence-defined ionic polypeptoid block copolymers consisting of a hydrophobic segment and a hydrophilic segment in an aqueous solution. By combining the results from MD simulations and experimental small-angle neutron scattering data, further insights were gained into the internal structure of the formed polypeptoid micelles, which is not always directly accessible from experiments. In addition, information was gained into the physics of the noncovalent interactions responsible for the self-assembly of weakly charged polypeptoids in an aqueous solution. While the aggregation number is governed by electrostatic repulsion of the negatively charged carboxylate (COO) substituents on the polypeptoid chain within the micelle, MD simulations indicate that the position of the charge on singly charged chains mediates the shape of the micelle through the charge-dipole interactions between the COO substituent and the surrounding water. Therefore, the polypeptoid micelles formed from the single-charged series offer the possibility for tailorable micelle shapes. In contrast, the polypeptoid micelles formed from the triple-charged series are characterized by more pronounced electrostatic repulsion that competes with more significant charge-sodium interactions, making it difficult to predict the shape of the micelles. This work has helped further develop design principles for the shape and structure of self-assembled micelles by controlling the position of charged moieties on the backbone of polypeptoid block copolymers.
静电相互作用在调节生物系统中起着重要作用,并且由于其在设计先进的刺激响应材料方面的实用性而受到越来越多的关注。聚肽是高度可调节的N-取代拟肽聚合物,缺乏主链氢键和手性。因此,聚肽是研究取代基非共价相互作用的影响而无主链分子内和分子间氢键复杂性的合适体系。在本研究中,对由一系列序列定义的离子型聚肽嵌段共聚物在水溶液中形成的胶束进行了全原子分子动力学(MD)模拟,该共聚物由疏水段和亲水段组成。通过结合MD模拟结果和实验小角中子散射数据,对形成的聚肽胶束的内部结构有了更深入的了解,这在实验中并不总是能直接获得的。此外,还获得了关于负责弱电荷聚肽在水溶液中自组装的非共价相互作用的物理信息。虽然聚集数由胶束内聚肽链上带负电的羧酸盐(COO)取代基的静电排斥作用决定,但MD模拟表明,单电荷链上电荷的位置通过COO取代基与周围水之间的电荷-偶极相互作用介导胶束的形状。因此,由单电荷系列形成的聚肽胶束提供了可定制胶束形状的可能性。相比之下,由三电荷系列形成的聚肽胶束的特点是静电排斥更明显,与更显著的电荷-钠相互作用相竞争,使得难以预测胶束的形状。这项工作通过控制聚肽嵌段共聚物主链上带电部分的位置,有助于进一步开发自组装胶束形状和结构的设计原则。