Department of Pharmaceutical Chemistry, Faculty of Pharmacy, The Islamia University of Bahawalpur, Bahawalpur 63100, Pakistan.
Department of Biochemistry, College of Science, King Saud University, P.O. Box 22452, Riyadh 11451, Saudi Arabia.
Molecules. 2022 Jun 25;27(13):4098. doi: 10.3390/molecules27134098.
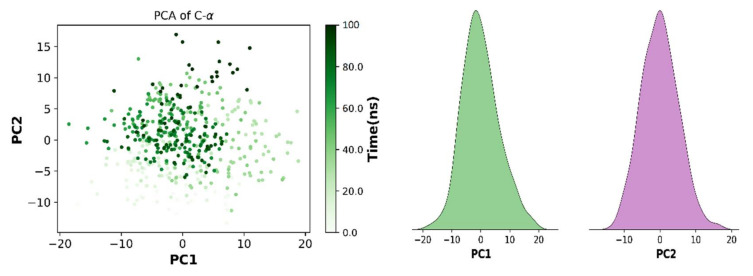
NIMA-related kinase7 (NEK7) plays a multifunctional role in cell division and NLRP3 inflammasone activation. A typical expression or any mutation in the genetic makeup of NEK7 leads to the development of cancer malignancies and fatal inflammatory disease, i.e., breast cancer, non-small cell lung cancer, gout, rheumatoid arthritis, and liver cirrhosis. Therefore, NEK7 is a promising target for drug development against various cancer malignancies. The combination of drug repurposing and structure-based virtual screening of large libraries of compounds has dramatically improved the development of anticancer drugs. The current study focused on the virtual screening of 1200 benzene sulphonamide derivatives retrieved from the PubChem database by selecting and docking validation of the crystal structure of NEK7 protein (PDB ID: 2WQN). The compounds library was subjected to virtual screening using Auto Dock Vina. The binding energies of screened compounds were compared to standard Dabrafenib. In particular, compound exhibited excellent binding energy of -42.67 kJ/mol, better than Dabrafenib (-33.89 kJ/mol). Selected drug candidates showed a reactive profile that was comparable to standard Dabrafenib. To characterize the stability of protein-ligand complexes, molecular dynamic simulations were performed, providing insight into the molecular interactions. The NEK7-Dabrafenib complex showed stability throughout the simulated trajectory. In addition, binding affinities, pIC50, and ADMET profiles of drug candidates were predicted using deep learning models. Deep learning models predicted the binding affinity of compound best among all derivatives, which supports the findings of virtual screening. These findings suggest that top hits can serve as potential inhibitors of NEK7. Moreover, it is recommended to explore the inhibitory potential of identified hits compounds through in-vitro and in-vivo approaches.
NIMA 相关激酶 7(NEK7)在细胞分裂和 NLRP3 炎症小体激活中发挥多种功能。NEK7 的典型表达或遗传组成中的任何突变都会导致癌症恶性肿瘤和致命炎症性疾病的发展,例如乳腺癌、非小细胞肺癌、痛风、类风湿性关节炎和肝硬化。因此,NEK7 是开发针对各种癌症恶性肿瘤的药物的有前途的靶标。药物再利用和基于结构的虚拟筛选大型化合物库的结合极大地促进了抗癌药物的开发。本研究侧重于通过选择和对接 NEK7 蛋白(PDB ID:2WQN)的晶体结构对从 PubChem 数据库中检索到的 1200 个苯磺酰胺衍生物进行虚拟筛选。使用 AutoDock Vina 对化合物库进行虚拟筛选。筛选化合物的结合能与标准 Dabrafenib 进行比较。特别是,化合物 表现出出色的结合能为-42.67 kJ/mol,优于 Dabrafenib(-33.89 kJ/mol)。所选药物候选物表现出与标准 Dabrafenib 相当的反应特征。为了表征蛋白质-配体复合物的稳定性,进行了分子动力学模拟,提供了对分子相互作用的深入了解。NEK7-Dabrafenib 复合物在整个模拟轨迹中表现出稳定性。此外,使用深度学习模型预测了候选药物的结合亲和力、pIC50 和 ADMET 谱。深度学习模型预测了所有衍生物中化合物 的结合亲和力最佳,这支持了虚拟筛选的结果。这些发现表明,顶级命中可能是 NEK7 的潜在抑制剂。此外,建议通过体外和体内方法探索鉴定出的命中化合物的抑制潜力。