Department of Integrative Biology, University of California, Berkeley, California, United States of America.
International laboratory of statistical and computational genomics, HSE University, Moscow, Russian Federation.
PLoS Genet. 2022 Jul 15;18(7):e1010281. doi: 10.1371/journal.pgen.1010281. eCollection 2022 Jul.
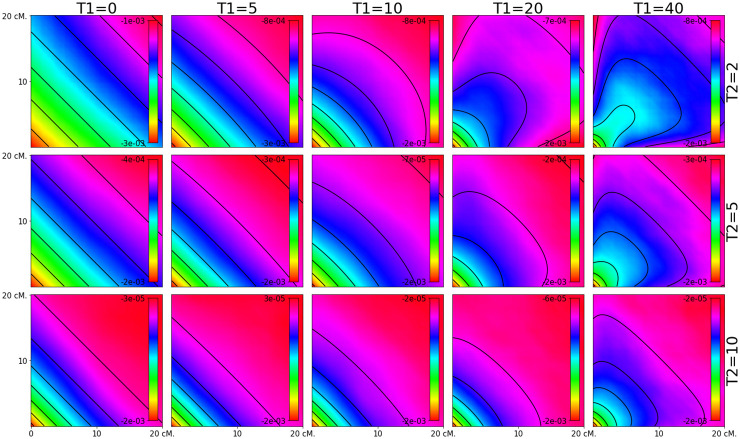
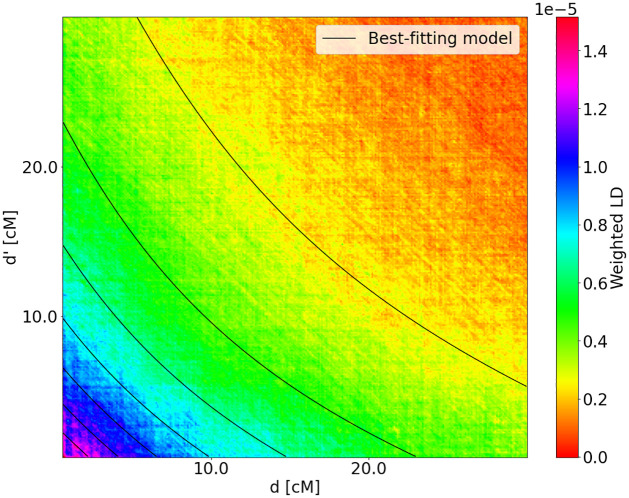
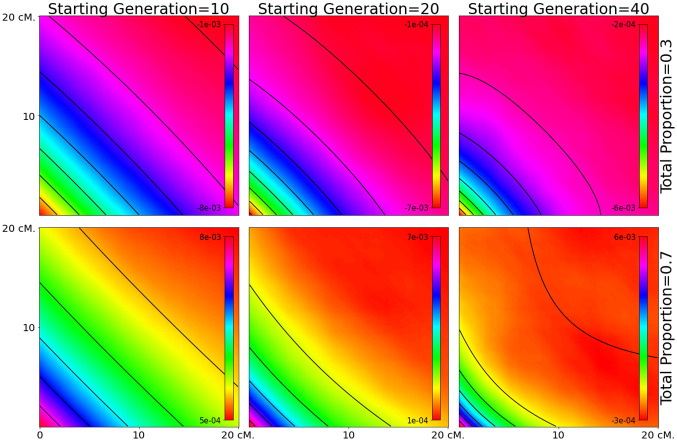
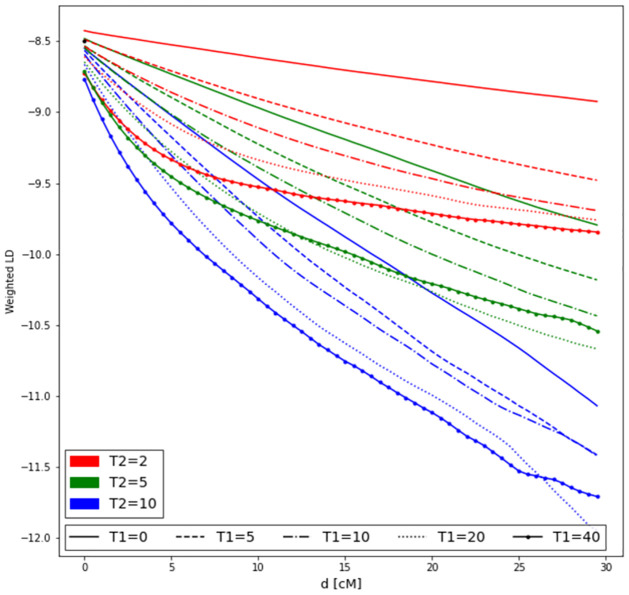
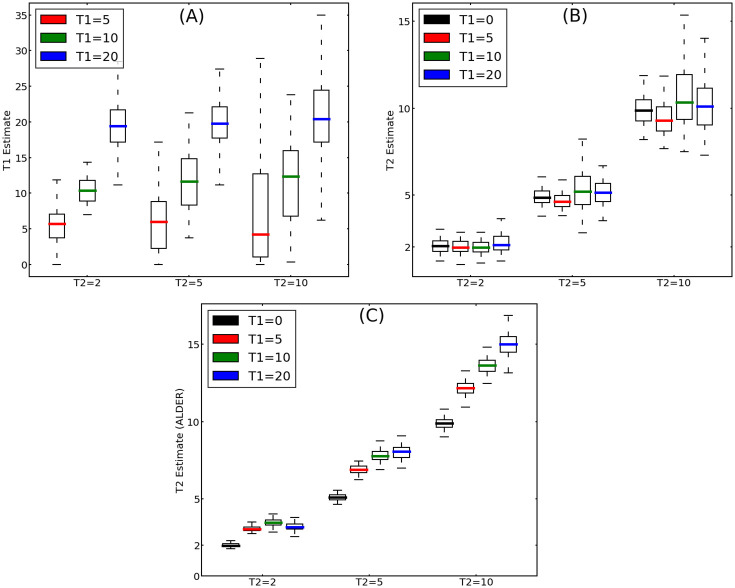
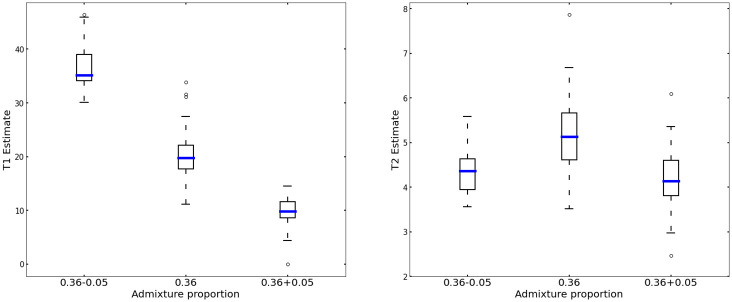
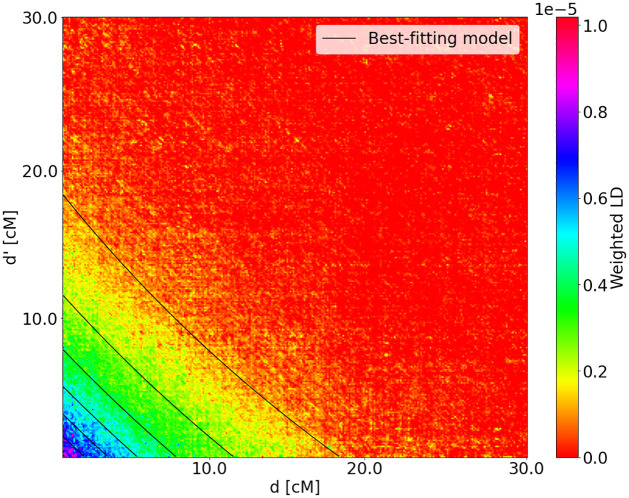
Estimating admixture histories is crucial for understanding the genetic diversity we see in present-day populations. Allele frequency or phylogeny-based methods are excellent for inferring the existence of admixture or its proportions. However, to estimate admixture times, spatial information from admixed chromosomes of local ancestry or the decay of admixture linkage disequilibrium (ALD) is used. One popular method, implemented in the programs ALDER and ROLLOFF, uses two-locus ALD to infer the time of a single admixture event, but is only able to estimate the time of the most recent admixture event based on this summary statistic. To address this limitation, we derive analytical expressions for the expected ALD in a three-locus system and provide a new statistical method based on these results that is able to resolve more complicated admixture histories. Using simulations, we evaluate the performance of this method on a range of different admixture histories. As an example, we apply the method to the Colombian and Mexican samples from the 1000 Genomes project. The implementation of our method is available at https://github.com/Genomics-HSE/LaNeta.
估算混合历史对于理解我们在当今人群中看到的遗传多样性至关重要。基于等位基因频率或系统发育的方法非常适合推断混合的存在或其比例。然而,要估计混合时间,需要使用来自混合染色体的局部亲缘关系的空间信息或混合连锁不平衡 (ALD) 的衰减。一种流行的方法,在程序 ALDER 和 ROLLOFF 中实现,使用两基因座 ALD 来推断单一混合事件的时间,但仅基于此汇总统计量能够估计最近一次混合事件的时间。为了解决这个限制,我们推导出了三基因座系统中预期的 ALD 的解析表达式,并基于这些结果提供了一种新的统计方法,该方法能够解决更复杂的混合历史。通过模拟,我们评估了该方法在一系列不同混合历史中的性能。作为一个例子,我们将该方法应用于 1000 基因组项目中的哥伦比亚和墨西哥样本。我们的方法的实现可在 https://github.com/Genomics-HSE/LaNeta 上获得。