Kelleher Jerome, Etheridge Alison M, McVean Gilean
Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom.
Department of Statistics, University of Oxford, Oxford, United Kingdom.
PLoS Comput Biol. 2016 May 4;12(5):e1004842. doi: 10.1371/journal.pcbi.1004842. eCollection 2016 May.
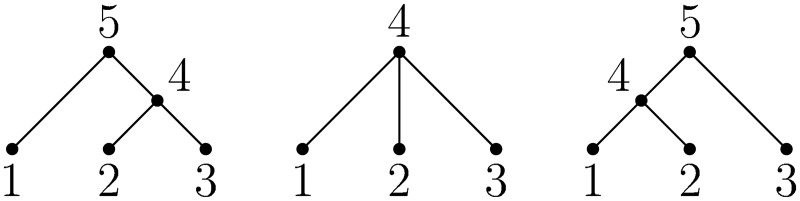
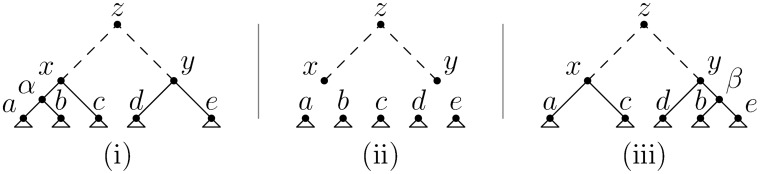
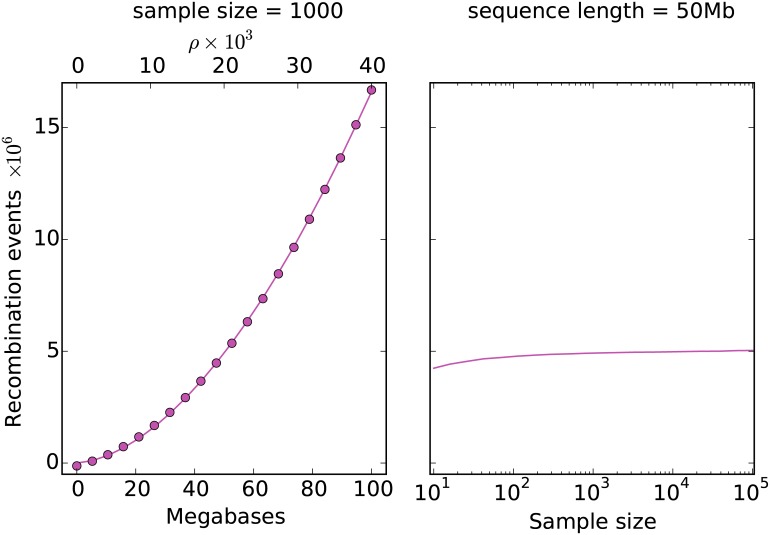
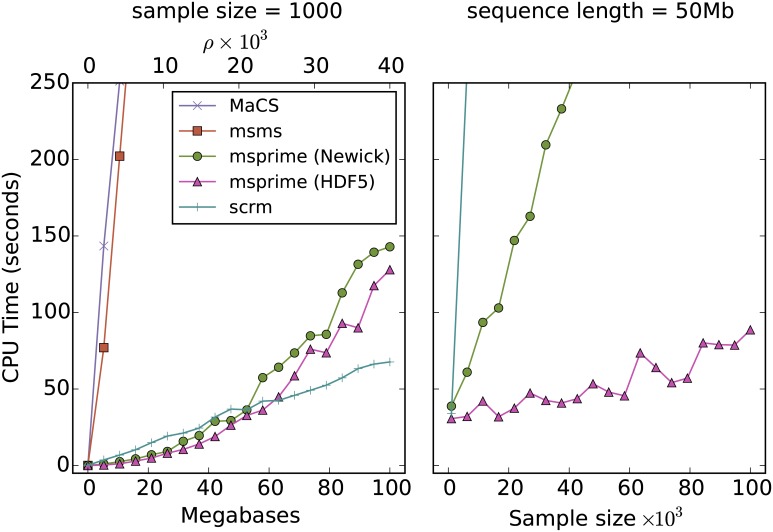
A central challenge in the analysis of genetic variation is to provide realistic genome simulation across millions of samples. Present day coalescent simulations do not scale well, or use approximations that fail to capture important long-range linkage properties. Analysing the results of simulations also presents a substantial challenge, as current methods to store genealogies consume a great deal of space, are slow to parse and do not take advantage of shared structure in correlated trees. We solve these problems by introducing sparse trees and coalescence records as the key units of genealogical analysis. Using these tools, exact simulation of the coalescent with recombination for chromosome-sized regions over hundreds of thousands of samples is possible, and substantially faster than present-day approximate methods. We can also analyse the results orders of magnitude more quickly than with existing methods.
基因变异分析中的一个核心挑战是对数百万个样本进行逼真的基因组模拟。目前的合并模拟扩展性不佳,或者使用的近似方法无法捕捉重要的长程连锁特性。分析模拟结果也面临重大挑战,因为当前存储系谱的方法占用大量空间、解析速度慢,且未利用相关树中的共享结构。我们通过引入稀疏树和合并记录作为系谱分析的关键单元来解决这些问题。使用这些工具,可以对数十万个样本的染色体大小区域进行带重组的合并精确模拟,且比目前的近似方法快得多。与现有方法相比,我们还能将结果分析速度提高几个数量级。