Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, Maryland.
Division of Pulmonary and Critical Care Medicine, Department of Medicine, University of Maryland School of Medicine, Baltimore, Maryland.
Am J Physiol Lung Cell Mol Physiol. 2022 Sep 1;323(3):L223-L239. doi: 10.1152/ajplung.00072.2022. Epub 2022 Jul 19.
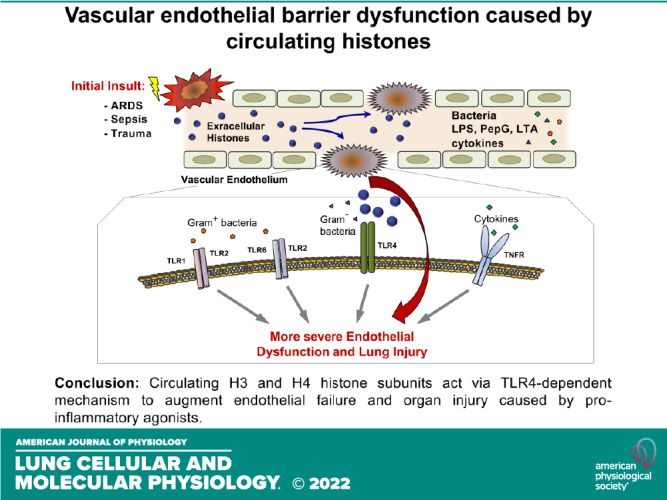
Extracellular histones released into the circulation following trauma, sepsis, and ARDS may act as potent damage-associated molecular pattern signals leading to multiple organ failure. Endothelial cell (EC) dysfunction caused by extracellular histones has been demonstrated in vitro and in vivo; however, precise mechanistic details of histone-induced EC dysfunction and exacerbation of ongoing inflammation remain poorly understood. This study investigated the role of extracellular histones in exacerbating preexisting endothelial dysfunction and acute lung injury. Histone subunits H3 and H4, but not H1, H2A, or H2B, induced permeability in human pulmonary EC. H3 and H4 at concentrations above 30 µg/mL caused EC inflammation reflected by activation of the NF-κB pathway, transcriptional activation, and release of cytokines and chemokines including IL-6 and IL-8, and increased mRNA and protein expression of EC adhesion molecules VCAM-1 and ICAM-1. Pharmacological inhibitors targeting Toll-like receptor TLR4 but not TLR2/6, blocked histone-induced EC dysfunction. H3 and H4 also strongly augmented EC permeability and inflammation caused by Gram-negative and Gram-positive bacterial particles, endotoxin, and TNFα. Heparin blocked histone-induced augmentation of EC inflammation caused by endotoxin and TNFα. Injection of histone in mouse models of lung injury caused by bacterial wall lipopolysaccharide (LPS) and heat-killed (HKSA) augmented ALI parameters: increased protein content, cell count, and inflammatory cytokine secretion in bronchoalveolar lavage fluid. Important clinical significance of these findings is in the demonstration that even a modest increase in extracellular histone levels can act as a severe exacerbating factor in conjunction with other EC barrier disruptive or proinflammatory agents.
创伤、脓毒症和 ARDS 后释放到循环中的细胞外组蛋白可作为潜在的损伤相关分子模式信号,导致多器官衰竭。已经在体外和体内证明了细胞外组蛋白引起的内皮细胞 (EC) 功能障碍;然而,组蛋白诱导的 EC 功能障碍和正在进行的炎症加剧的确切机制细节仍知之甚少。本研究调查了细胞外组蛋白在加剧预先存在的内皮功能障碍和急性肺损伤中的作用。组蛋白亚基 H3 和 H4,但不是 H1、H2A 或 H2B,诱导人肺 EC 的通透性。浓度高于 30μg/mL 的 H3 和 H4 引起 EC 炎症,反映为 NF-κB 途径激活、转录激活以及细胞因子和趋化因子(包括 IL-6 和 IL-8)的释放,以及 EC 粘附分子 VCAM-1 和 ICAM-1 的 mRNA 和蛋白表达增加。针对 Toll 样受体 TLR4 的药理学抑制剂,但不是 TLR2/6,阻断了组蛋白诱导的 EC 功能障碍。H3 和 H4 还强烈增强了革兰氏阴性和革兰氏阳性细菌颗粒、内毒素和 TNFα 引起的 EC 通透性和炎症。肝素阻断了内毒素和 TNFα 引起的组蛋白诱导的 EC 炎症的增强。在细菌细胞壁脂多糖 (LPS) 和热灭活 (HKSA) 引起的肺损伤小鼠模型中注射组蛋白,增加了蛋白质含量、细胞计数和支气管肺泡灌洗液中炎症细胞因子的分泌。这些发现的重要临床意义在于证明,即使细胞外组蛋白水平略有增加,也可以与其他破坏 EC 屏障或促炎的药物一起作为严重的加剧因素。