Alkhzem Abdulaziz H, Woodman Timothy J, Blagbrough Ian S
Department of Pharmacy and Pharmacology, University of Bath Bath BA2 7AY UK
RSC Adv. 2022 Jul 6;12(30):19470-19484. doi: 10.1039/d2ra03281c. eCollection 2022 Jun 29.
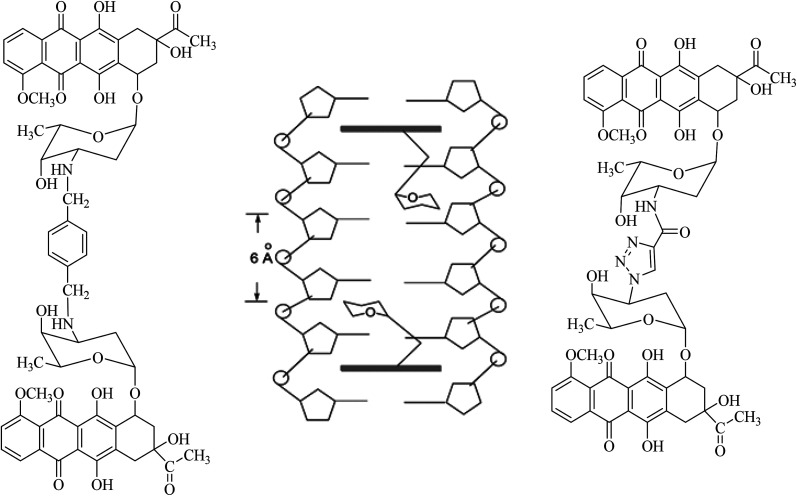
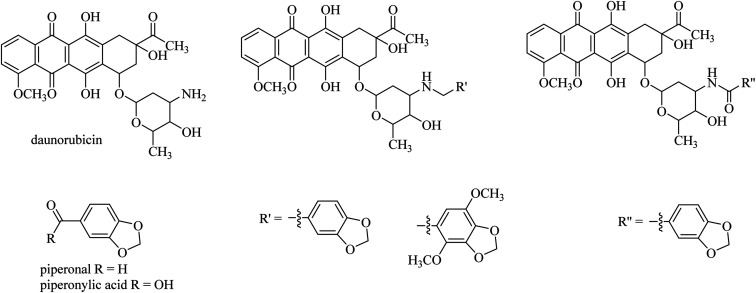
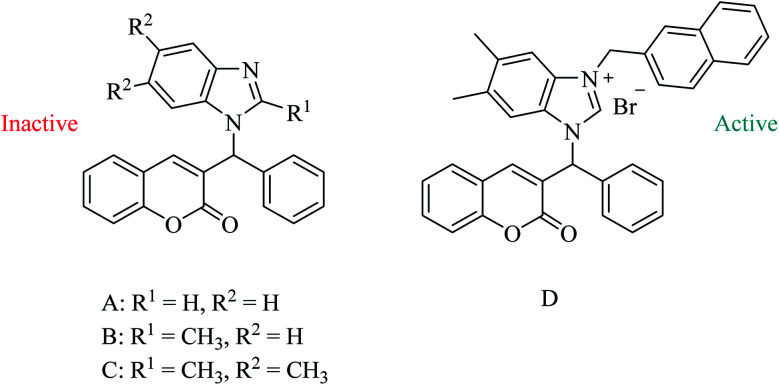
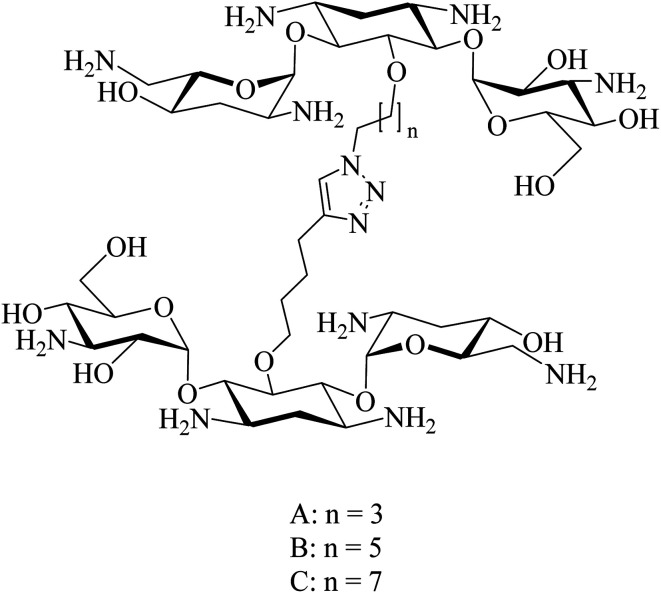
The development of highly effective conjugate chemistry approaches is a way to improve the quality of drugs and of medicines. The aim of this paper is to highlight and review such hybrid compounds and the strategies underpinning their design. A variety of unique hybrid compounds provide an excellent toolkit for novel biological activity, anticancer and non-viral gene therapy (NVGT), and as templates for killing bacteria and preventing antibiotic drug resistance. First we discuss the anticancer potential of hybrid compounds, containing daunorubicin, benzyl- or tetrahydroisoquinoline-coumarin, and cytotoxic NSAID-pyrrolizidine/indolizine hybrids, then NVGT cationic lipid-based delivery agents, where steroids or long chain fatty acids as the lipid moiety are bound to polyamines as the cationic moiety. These polyamines can be linear as in spermidine or spermine, or on a polycyclic sugar template, aminoglycosides kanamycin and neomycin B, the latter substituted with six amino groups. They are highly efficient for the delivery of both fluorescent DNA and siRNA. Molecular precedents can be found for the design of hybrid compounds in the natural world, , squalamine, the first representative of a previously unknown class of natural antibiotics of animal origin. These polyamine-bile acid ( cholic acid type) conjugates display many exciting biological activities with the bile acids acting as a lipidic region and spermidine as the polycationic region. Analogues of squalamine can act as vectors in NVGT. Their natural role is as antibiotics. Novel antibacterial materials are urgently needed as recalcitrant bacterial infection is a worldwide problem for human health. Ribosome inhibitors founded upon dimers of tobramycin or neomycin, bound as ethers by a 1,6-hexyl linker or a more complex diether-disulfide linker, improved upon the antibiotic activity of aminoglycoside monomers by 20- to 1200-fold. Other hybrids, linked by click chemistry, conjugated ciprofloxacin to neomycin, trimethoprim, or tedizolid, which is now in clinical trials.
开发高效的共轭化学方法是提高药物和药品质量的一种途径。本文的目的是突出并综述这类杂合化合物及其设计所依据的策略。多种独特的杂合化合物为新型生物活性、抗癌和非病毒基因治疗(NVGT)提供了出色的工具包,并且可作为杀灭细菌和预防抗生素耐药性的模板。首先,我们讨论含柔红霉素、苄基或四氢异喹啉 - 香豆素的杂合化合物以及细胞毒性非甾体抗炎药 - 吡咯里西啶/中氮茚杂合物的抗癌潜力,然后讨论基于NVGT阳离子脂质的递送剂,其中作为脂质部分的类固醇或长链脂肪酸与作为阳离子部分的多胺结合。这些多胺可以是亚精胺或精胺那样的线性结构,或者基于多环糖模板,如氨基糖苷类的卡那霉素和新霉素B,后者带有六个氨基。它们对于荧光DNA和小干扰RNA(siRNA)的递送都非常高效。在自然界中可以找到杂合化合物设计的分子先例,如鲨胺,它是动物源一类此前未知的天然抗生素的首个代表。这些多胺 - 胆汁酸(胆酸类型)共轭物展现出许多令人兴奋的生物活性,其中胆汁酸作为脂质区域,亚精胺作为聚阳离子区域。鲨胺类似物可在NVGT中充当载体。它们的天然作用是作为抗生素。由于顽固的细菌感染是全球人类健康面临的问题,因此迫切需要新型抗菌材料。基于妥布霉素或新霉素二聚体的核糖体抑制剂,通过1,6 - 己基连接基或更复杂的二醚 - 二硫连接基作为醚键连接,其抗生素活性比氨基糖苷单体提高了20至1200倍。其他通过点击化学连接的杂合物,将环丙沙星与新霉素、甲氧苄啶或替地唑胺共轭,替地唑胺目前正在进行临床试验。