Division of Cardiovascular Medicine, Department of Internal Medicine, University of Iowa Carver College of Medicine, Iowa City, Iowa, USA.
Abboud Cardiovascular Research Center, University of Iowa Carver College of Medicine, Iowa City, Iowa, USA.
Clin Transl Med. 2022 Jul;12(7):e954. doi: 10.1002/ctm2.954.
Mice with deletion of complex I subunit Ndufs4 develop mitochondrial encephalomyopathy resembling Leigh syndrome (LS). The metabolic derangement and underlying mechanisms of cardio-encephalomyopathy in LS remains incompletely understood.
We performed echocardiography, electrophysiology, confocal microscopy, metabolic and molecular/morphometric analysis of the mice lacking Ndufs4. HEK293 cells, human iPS cells-derived cardiomyocytes and neurons were used to determine the mechanistic role of mitochondrial complex I deficiency.
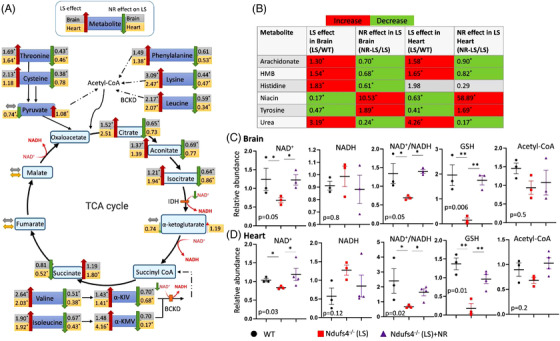
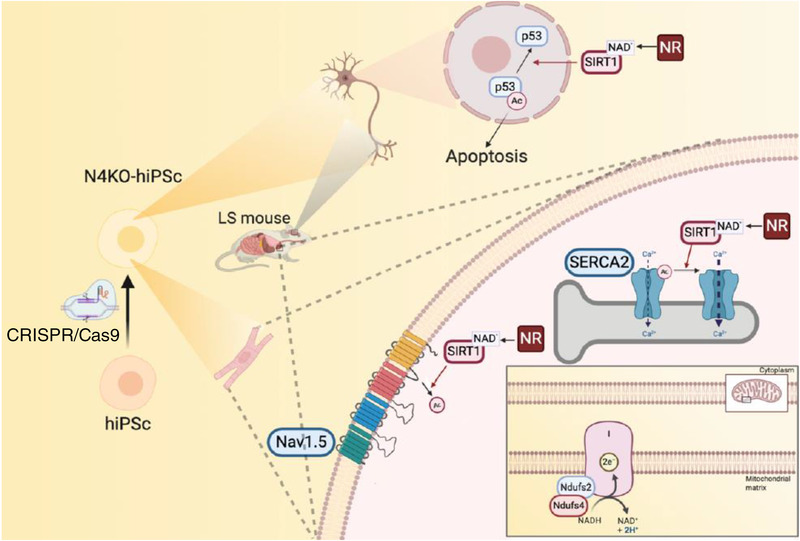
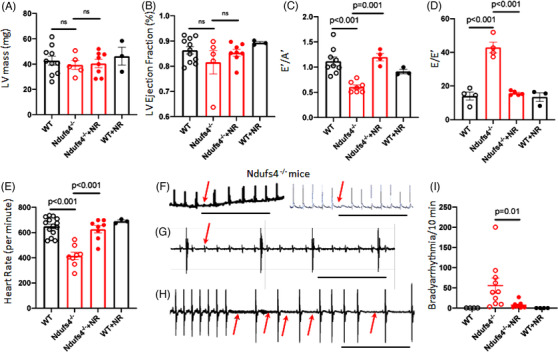
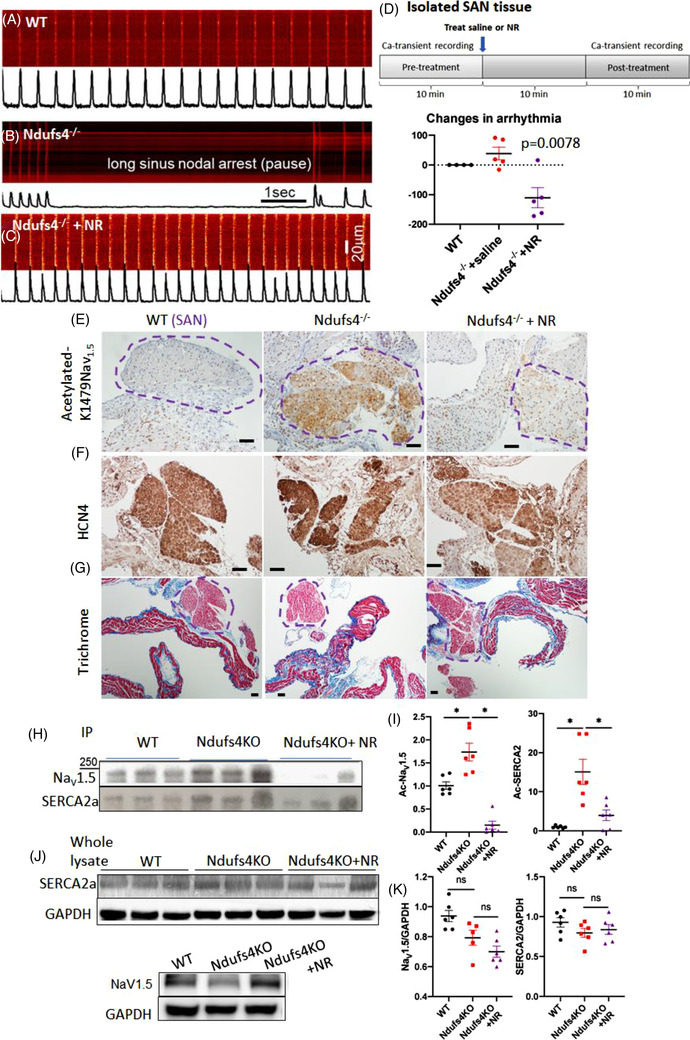
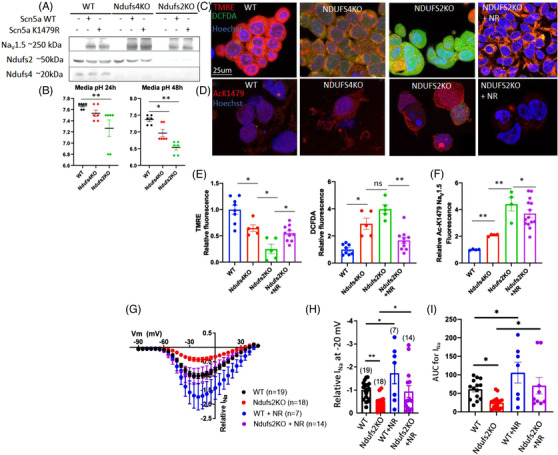
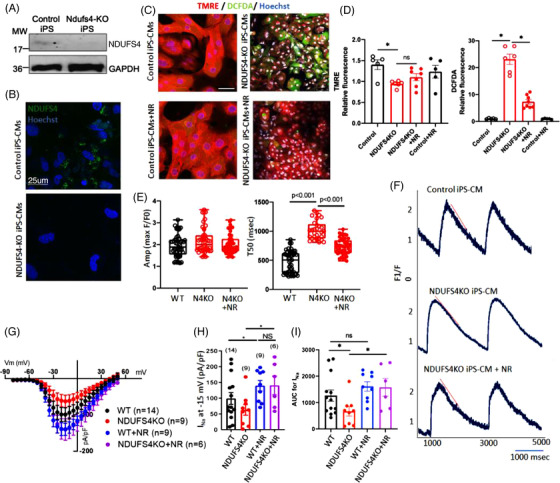
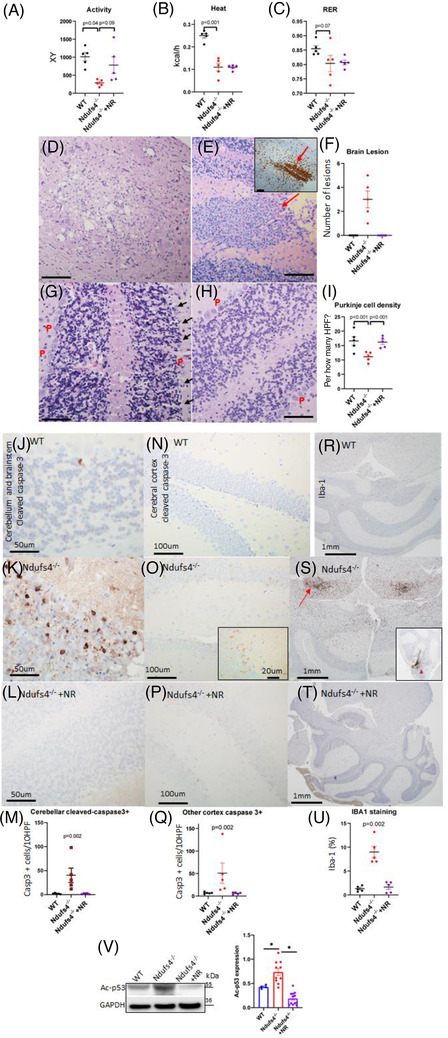
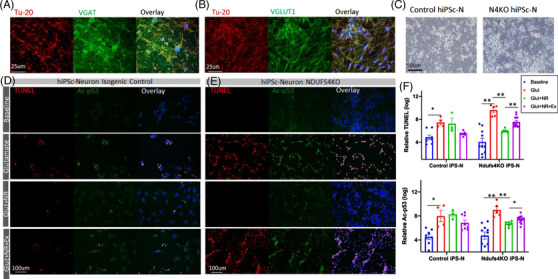
LS mice develop severe cardiac bradyarrhythmia and diastolic dysfunction. Human-induced pluripotent stem cell-derived cardiomyocytes (iPS-CMs) with Ndufs4 deletion recapitulate LS cardiomyopathy. Mechanistically, we demonstrate a direct link between complex I deficiency, decreased intracellular (nicotinamide adenine dinucleotide) NAD /NADH and bradyarrhythmia, mediated by hyperacetylation of the cardiac sodium channel Na 1.5, particularly at K1479 site. Neuronal apoptosis in the cerebellar and midbrain regions in LS mice was associated with hyperacetylation of p53 and activation of microglia. Targeted metabolomics revealed increases in several amino acids and citric acid cycle intermediates, likely due to impairment of NAD -dependent dehydrogenases, and a substantial decrease in reduced Glutathione (GSH). Metabolic rescue by nicotinamide riboside (NR) supplementation increased intracellular NAD / NADH, restored metabolic derangement, reversed protein hyperacetylation through NAD -dependent Sirtuin deacetylase, and ameliorated cardiomyopathic phenotypes, concomitant with improvement of Na 1.5 current and SERCA2a function measured by Ca2 -transients. NR also attenuated neuronal apoptosis and microglial activation in the LS brain and human iPS-derived neurons with Ndufs4 deletion.
Our study reveals direct mechanistic explanations of the observed cardiac bradyarrhythmia, diastolic dysfunction and neuronal apoptosis in mouse and human induced pluripotent stem cells (iPSC) models of LS.
缺失复合体 I 亚基 Ndufs4 的小鼠会发展出类似于 Leigh 综合征(LS)的线粒体脑肌病。LS 中心-脑肌病的代谢紊乱和潜在机制仍不完全清楚。
我们对缺乏 Ndufs4 的小鼠进行了超声心动图、电生理学、共聚焦显微镜、代谢和分子/形态计量学分析。使用 HEK293 细胞、人诱导多能干细胞(iPS)衍生的心肌细胞和神经元来确定线粒体复合体 I 缺陷的机制作用。
LS 小鼠出现严重的心脏心动过缓伴舒张功能障碍。缺乏 Ndufs4 的人诱导多能干细胞衍生的心肌细胞(iPS-CMs)可再现 LS 心肌病。从机制上讲,我们证明了复合体 I 缺陷、细胞内(烟酰胺腺嘌呤二核苷酸)NAD/NADH 减少与心脏钠离子通道 Na 1.5 的心动过缓之间存在直接联系,这种联系是通过心脏钠离子通道 Na 1.5 的乙酰化过度介导的,特别是在 K1479 位点。LS 小鼠小脑和中脑区域的神经元凋亡与 p53 的乙酰化过度和小胶质细胞的激活有关。靶向代谢组学显示,几种氨基酸和柠檬酸循环中间产物增加,可能是由于 NAD 依赖性脱氢酶的损伤,以及还原型谷胱甘肽(GSH)的大量减少。烟酰胺核糖苷(NR)补充的代谢挽救增加了细胞内 NAD/NADH,恢复了代谢紊乱,通过 NAD 依赖性 Sirtuin 去乙酰化酶逆转了蛋白质的过度乙酰化,并改善了心肌病表型,同时改善了通过 Ca2-瞬变测量的 Na 1.5 电流和 SERCA2a 功能。NR 还减轻了 LS 大脑中的神经元凋亡和小胶质细胞激活,以及缺乏 Ndufs4 的人诱导多能干细胞衍生神经元中的神经元凋亡和小胶质细胞激活。
我们的研究揭示了 LS 小鼠和人诱导多能干细胞(iPSC)模型中观察到的心脏心动过缓、舒张功能障碍和神经元凋亡的直接机制解释。