Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA.
Department of Biology and Huck Institute of the Life Sciences, The Pennsylvania State University, University Park, PA, USA.
Nat Commun. 2022 Jul 25;13(1):4312. doi: 10.1038/s41467-022-31872-6.
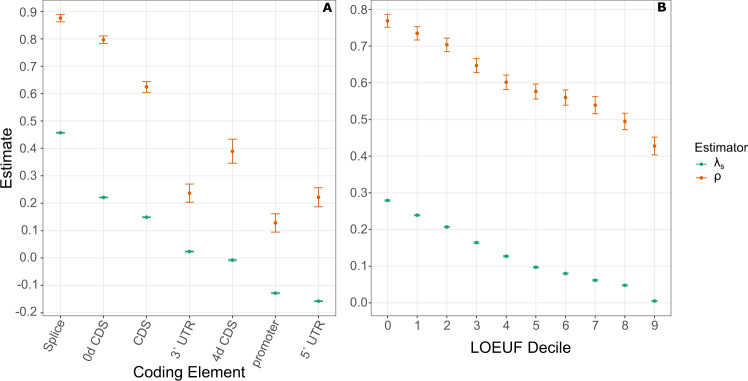
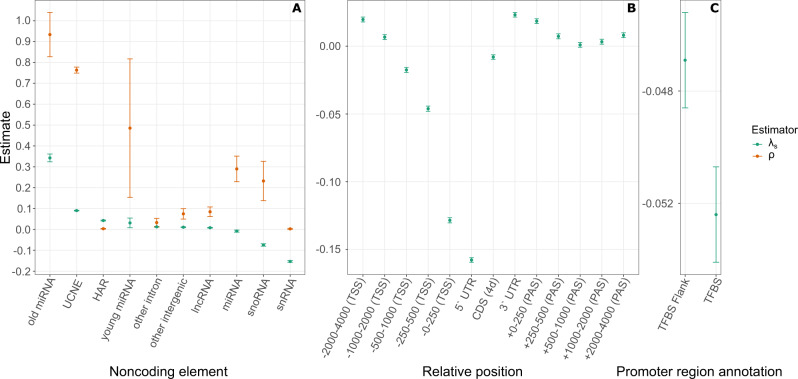
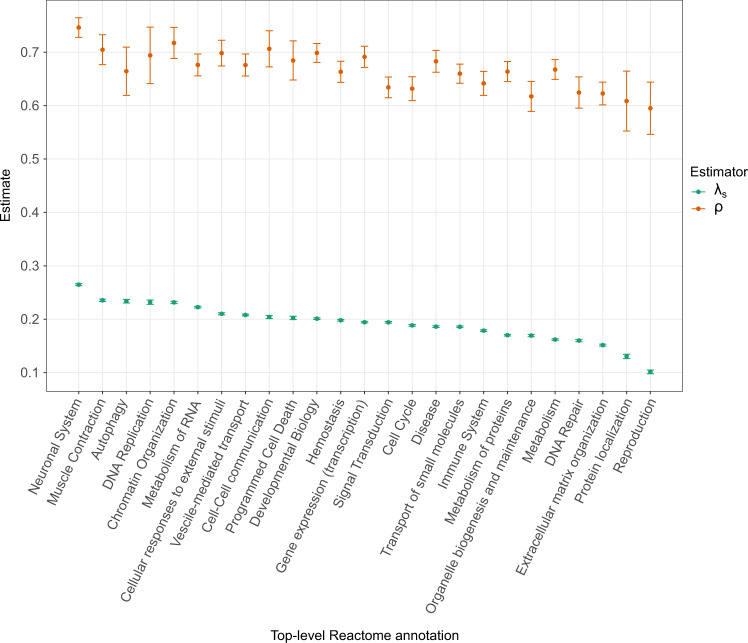
Large-scale genome sequencing has enabled the measurement of strong purifying selection in protein-coding genes. Here we describe a new method, called ExtRaINSIGHT, for measuring such selection in noncoding as well as coding regions of the human genome. ExtRaINSIGHT estimates the prevalence of "ultraselection" by the fractional depletion of rare single-nucleotide variants, after controlling for variation in mutation rates. Applying ExtRaINSIGHT to 71,702 whole genome sequences from gnomAD v3, we find abundant ultraselection in evolutionarily ancient miRNAs and neuronal protein-coding genes, as well as at splice sites. By contrast, we find much less ultraselection in other noncoding RNAs and transcription factor binding sites, and only modest levels in ultraconserved elements. We estimate that ~0.4-0.7% of the human genome is ultraselected, implying ~ 0.26-0.51 strongly deleterious mutations per generation. Overall, our study sheds new light on the genome-wide distribution of fitness effects by combining deep sequencing data and classical theory from population genetics.
大规模基因组测序使我们能够测量蛋白质编码基因中的强纯化选择。在这里,我们描述了一种新的方法,称为 ExtRaINSIGHT,用于测量人类基因组中非编码区和编码区的这种选择。ExtRaINSIGHT 通过控制突变率的变化来估计稀有单核苷酸变异的分数耗竭,从而估计“超选择”的流行程度。将 ExtRaINSIGHT 应用于 gnomAD v3 中的 71,702 个全基因组序列,我们发现进化古老的 miRNA 和神经元蛋白编码基因以及剪接位点存在丰富的超选择。相比之下,我们在其他非编码 RNA 和转录因子结合位点中发现的超选择要少得多,在超保守元件中只发现适度的超选择。我们估计人类基因组中有 0.4-0.7%是超选择的,这意味着每代有 0.26-0.51 个强有害突变。总的来说,我们的研究通过结合深度测序数据和来自群体遗传学的经典理论,为了解全基因组适应度效应的分布提供了新的视角。