Graduate Field of Computer Science, Cornell University, Ithaca, NY, USA.
Simons Center for Quantitative Biology, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA.
Nat Genet. 2019 Feb;51(2):335-342. doi: 10.1038/s41588-018-0300-z. Epub 2018 Dec 17.
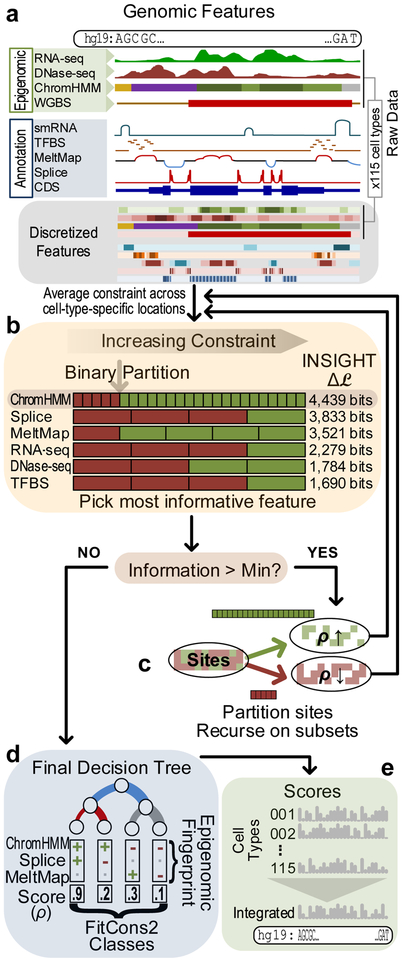
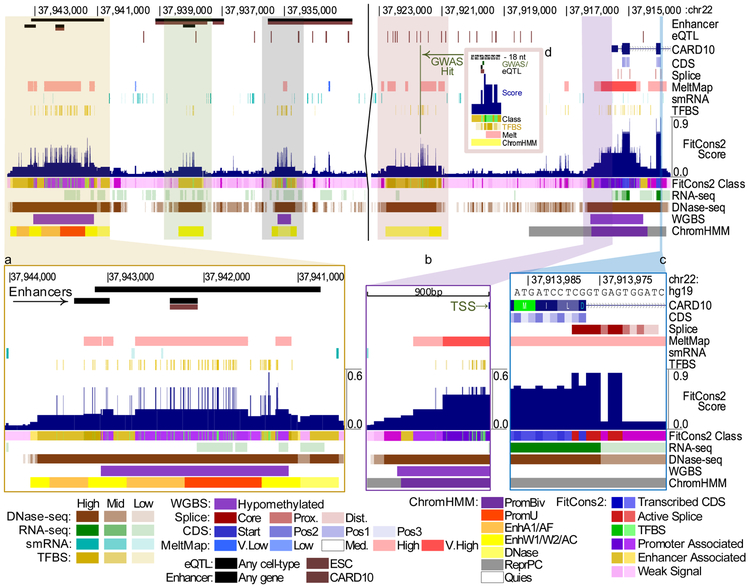
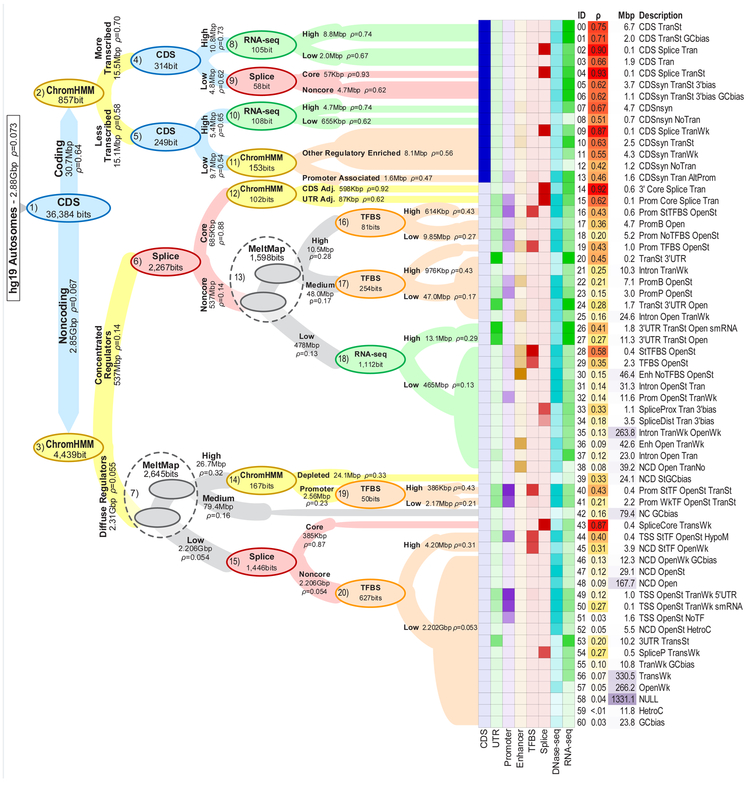
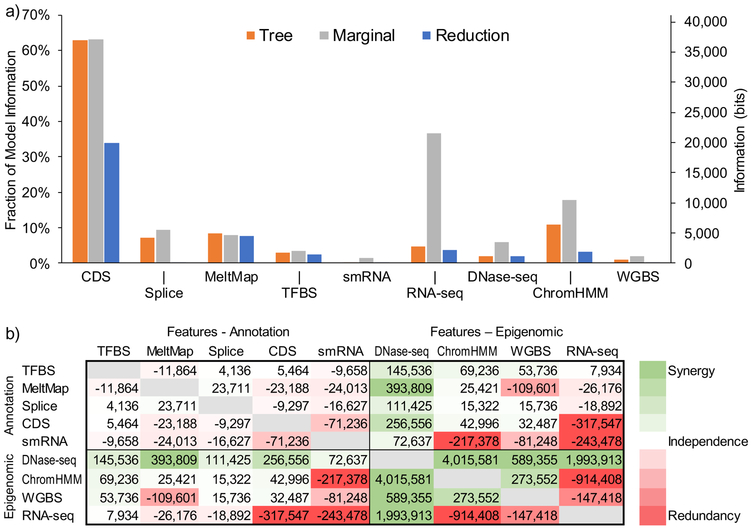
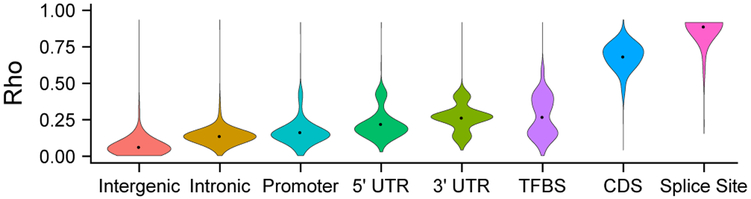
Here we ask the question "How much information do epigenomic datasets provide about human genomic function?" We consider nine epigenomic features across 115 cell types and measure information about function as a reduction in entropy under a probabilistic evolutionary model fitted to human and nonhuman primate genomes. Several epigenomic features yield more information in combination than they do individually. We find that the entropy in human genetic variation predominantly reflects a balance between mutation and neutral drift. Our cell-type-specific FitCons scores reveal relationships among cell types and suggest that around 8% of nucleotide sites are constrained by natural selection.
在这里,我们提出了一个问题:“表观基因组数据集提供了多少关于人类基因组功能的信息?”我们考虑了 115 种细胞类型中的九个表观基因组特征,并通过拟合人类和非人类灵长类基因组的概率进化模型,将功能信息衡量为熵的减少。几个表观基因组特征的组合比单独使用时提供了更多的信息。我们发现,人类遗传变异中的熵主要反映了突变和中性漂变之间的平衡。我们的细胞类型特异性 FitCons 评分揭示了细胞类型之间的关系,并表明大约 8%的核苷酸位点受到自然选择的限制。