Department of Biological Sciences, Kongju National University, Gongju 32588, Korea.
Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul 06351, Korea.
Genes (Basel). 2022 Jul 8;13(7):1219. doi: 10.3390/genes13071219.
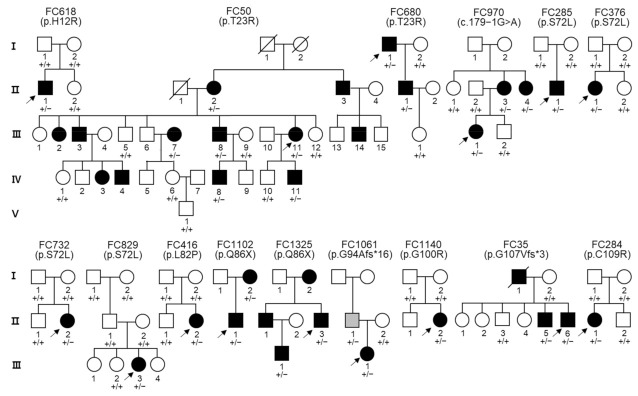
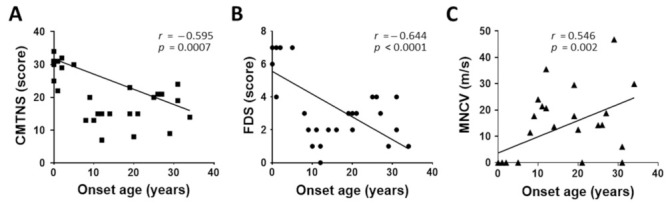
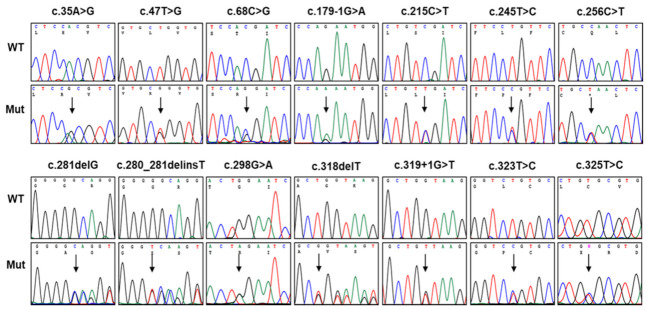
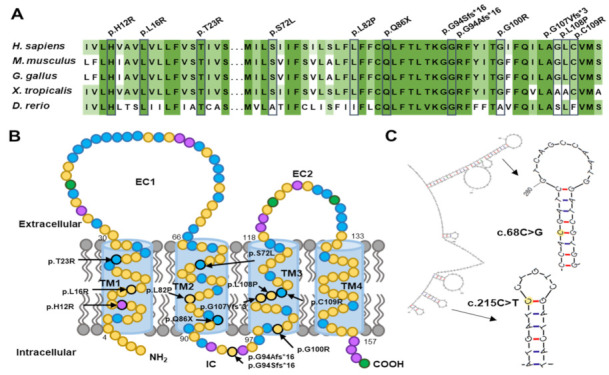
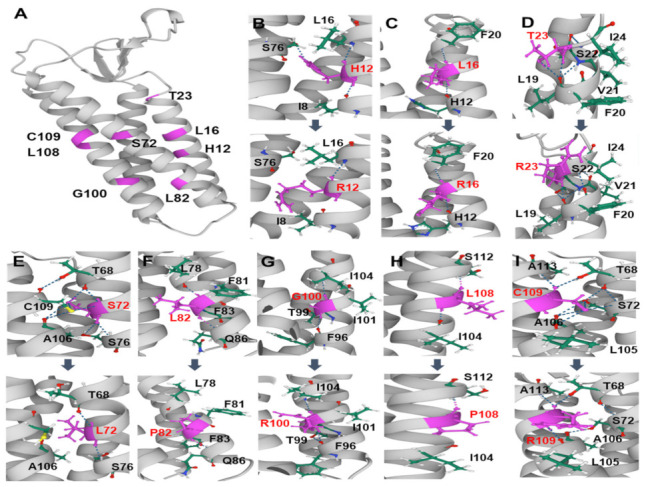
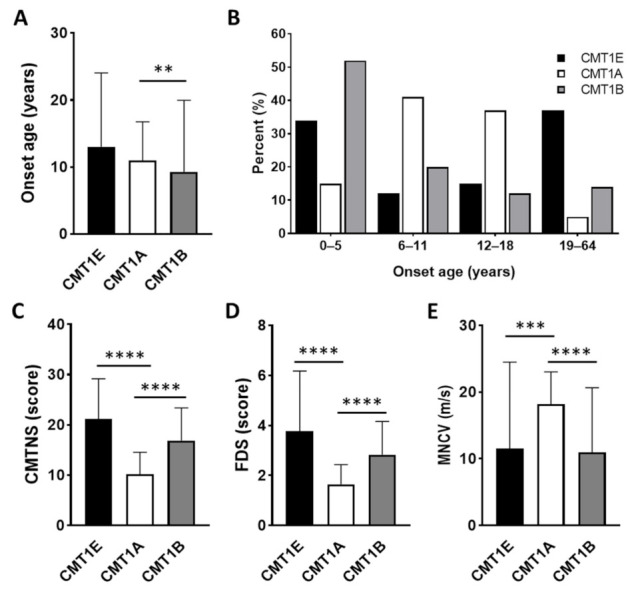
Duplication and deletion of the peripheral myelin protein 22 (PMP22) gene cause Charcot-Marie-Tooth disease type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP), respectively, while point mutations or small insertions and deletions (indels) usually cause CMT type 1E (CMT1E) or HNPP. This study was performed to identify PMP22 mutations and to analyze the genotype−phenotype correlation in Korean CMT families. By the application of whole-exome sequencing (WES) and targeted gene panel sequencing (TS), we identified 14 pathogenic or likely pathogenic PMP22 mutations in 21 families out of 850 CMT families who were negative for 17p12 (PMP22) duplication. Most mutations were located in the well-conserved transmembrane domains. Of these, eight mutations were not reported in other populations. High frequencies of de novo mutations were observed, and the mutation sites of c.68C>G and c.215C>T were suggested as the mutational hotspots. Affected individuals showed an early onset-severe phenotype and late onset-mild phenotype, and more than 40% of the CMT1E patients showed hearing loss. Physical and electrophysiological symptoms of the CMT1E patients were more severely damaged than those of CMT1A while similar to CMT1B caused by MPZ mutations. Our results will be useful for the reference data of Korean CMT1E and the molecular diagnosis of CMT1 with or without hearing loss.
周围髓鞘蛋白 22(PMP22)基因的重复和缺失分别导致 Charcot-Marie-Tooth 病 1A 型(CMT1A)和遗传性压力易发性神经病(HNPP),而点突变或小插入和缺失(indels)通常导致 CMT 型 1E(CMT1E)或 HNPP。本研究旨在鉴定 PMP22 突变,并分析韩国 CMT 家族的基因型-表型相关性。通过全外显子组测序(WES)和靶向基因panel 测序(TS),我们在 850 个 CMT 家族中,有 21 个家族排除了 17p12(PMP22)重复,发现了 14 个致病性或可能致病性的 PMP22 突变。大多数突变位于高度保守的跨膜结构域。其中,有 8 个突变在其他人群中未报道过。我们观察到高频的新生突变,c.68C>G 和 c.215C>T 的突变位点被认为是突变热点。受影响的个体表现出早发-严重表型和晚发-轻度表型,超过 40%的 CMT1E 患者有听力损失。CMT1E 患者的身体和电生理症状比 CMT1A 患者更严重,但与 MPZ 突变引起的 CMT1B 相似。我们的结果将为韩国 CMT1E 的参考数据和有无听力损失的 CMT1 的分子诊断提供有用的信息。