Department of Pathology, Assistance Publique - Hôpitaux de Paris, Henri-Mondor Hospital, 94010, Créteil, France.
INSERM U955 Institut Mondor de Recherche Biomédicale (IMRB), Paris Est Créteil University, 94010, Créteil, France.
Br J Dermatol. 2022 Dec;187(6):970-980. doi: 10.1111/bjd.21791. Epub 2022 Sep 2.
Primary cutaneous peripheral T-cell lymphomas with a T-follicular helper phenotype (pcTFH-PTCL) are poorly characterized, and often compared to, but not corresponding with, mycosis fungoides (MF), Sézary syndrome, primary cutaneous CD4 lymphoproliferative disorder, and skin manifestations of angioimmunoblastic T-cell lymphomas (AITL).
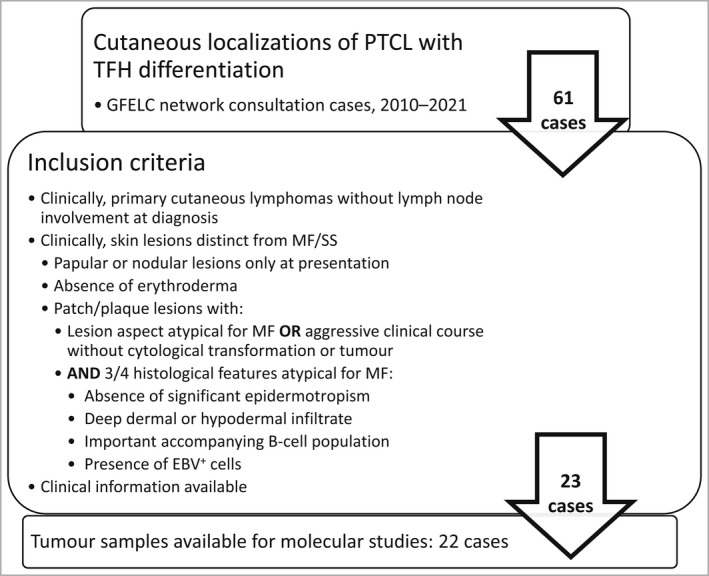
We describe the clinicopathological features of pcTFH-PTCL in this original series of 23 patients, and also characterize these cases molecularly.
Clinical and histopathological data of the selected patients were reviewed. Patient biopsy samples were also analysed by targeted next-generation sequencing.
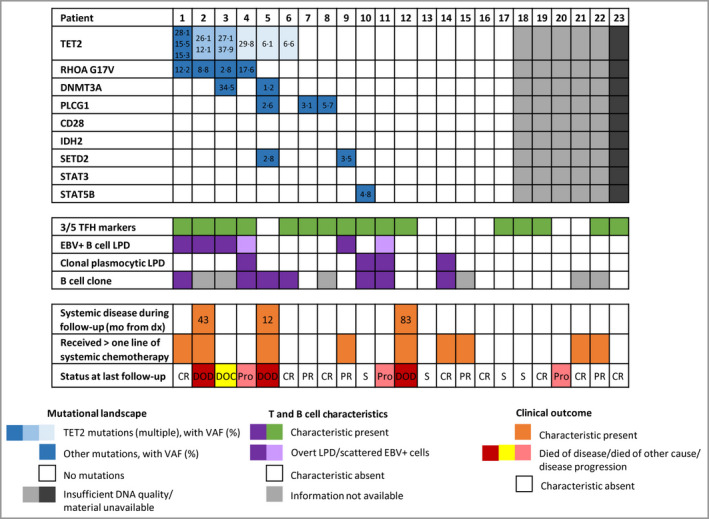
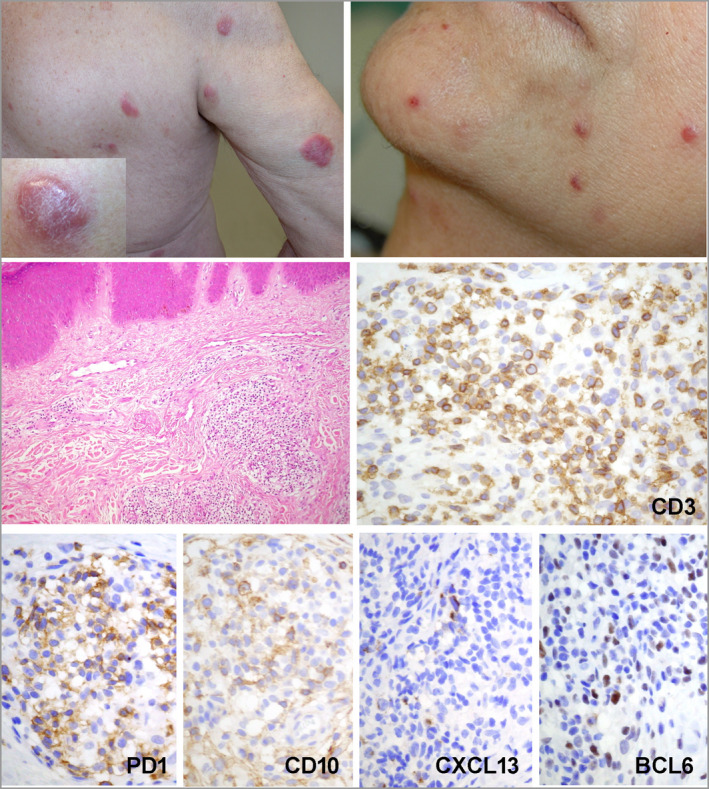
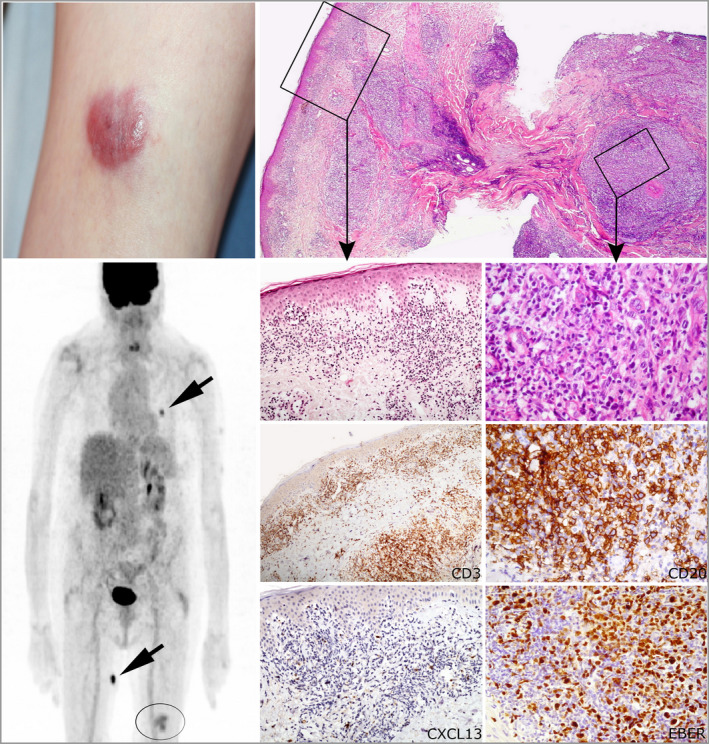
All patients (15 men, eight women; median age 66 years) presented with skin lesions, without systemic disease. Most were stage T3b, with nodular (n = 16), papular (n = 6) or plaque (atypical for MF, n = 1) lesions. Three (13%) developed systemic disease and died of lymphoma. Nine (39%) patients received more than one line of chemotherapy. Histologically, the lymphomas were CD4 T-cell proliferations, usually dense and located in the deep dermis (n = 14, 61%), with the expression of at least two TFH markers (CD10, CXCL13, PD1, ICOS, BCL6), including three markers in 16 cases (70%). They were associated with a variable proportion of B cells. Eight patients were diagnosed with an associated B-cell lymphoproliferative disorder (LPD) on biopsy, including Epstein-Barr virus (EBV)-positive diffuse large B-cell lymphoma (n = 3), EBV LPD (n = 1) and monotypic plasma cell LPD (n = 4). Targeted sequencing showed four patients to have a mutated TET2-RHOAG17V association (as frequently seen in AITL) and another a TET2/DNMT3A/PLCG1/SETD2 mutational profile. The latter patient, one with a TET2-RHOA association, and one with no detected mutations, developed systemic disease and died. Five other patients showed isolated mutations in TET2 (n = 1), PLCG1 (n = 2), SETD2 (n = 1) or STAT5B (n = 1).
Patients with pcTFH-PTCL have pathological and genetic features that overlap with those of systemic lymphoma of TFH derivation. Clinically, most remained confined to the skin, with only three patients showing systemic spread and death. Whether pcTFH-PTCL should be integrated as a new subgroup of TFH lymphomas in future classifications is still a matter of debate. What is already known about this topic? There is a group of cutaneous lymphomas that express T-follicular helper (TFH) markers that do not appear to correspond to existing World Health Organization diagnostic entities. These include mycosis fungoides, Sézary syndrome, or primary cutaneous CD4 small/medium-sized T-cell lymphoproliferative disorder or cutaneous extensions of systemic peripheral T-cell lymphomas (PTCL) with TFH phenotype. What does this study add? This is the first large original series of patients with a diagnosis of primary cutaneous PTCL with a TFH phenotype (pcTFH-PTCL) to be molecularly characterized. pcTFH-PTCL may be a standalone group of cutaneous lymphomas with clinicopathological and molecular characteristics that overlap with those of systemic TFH lymphomas, such as angioimmunoblastic T-cell lymphoma, and does not belong to known diagnostic groups of cutaneous lymphoma. This has an impact on the treatment and follow-up of patients; the clinical behaviour needs to be better clarified in further studies to tailor patient management.
原发性皮肤外周 T 细胞淋巴瘤伴 T 滤泡辅助表型(pcTFH-PTCL)的特征尚未完全明确,常与蕈样真菌病(MF)、Sézary 综合征、原发性皮肤 CD4 淋巴增生性疾病和血管免疫母细胞性 T 细胞淋巴瘤(AITL)的皮肤表现相比较,但并不完全对应。
我们描述了 23 例原发性 pcTFH-PTCL 患者的临床病理特征,并对这些病例进行了分子特征分析。
回顾了所选患者的临床和组织病理学数据。还对患者活检样本进行了靶向二代测序分析。
所有患者(15 名男性,8 名女性;中位年龄 66 岁)均有皮肤病变,无全身疾病。大多数患者为 T3b 期,表现为结节性(n=16)、丘疹性(n=6)或斑块性(非典型 MF,n=1)病变。3 例(13%)出现全身疾病并死于淋巴瘤。9 例(39%)患者接受了超过一线的化疗。组织学上,这些淋巴瘤为 CD4 T 细胞增生,通常密集位于真皮深部(n=14,61%),表达至少两种 TFH 标志物(CD10、CXCL13、PD1、ICOS、BCL6),其中 16 例(70%)表达了三种标志物。它们与一定比例的 B 细胞相关。8 例患者在活检时被诊断为伴发 B 细胞淋巴增生性疾病(LPD),包括 EBV 阳性弥漫性大 B 细胞淋巴瘤(n=3)、EBV LPD(n=1)和单克隆浆细胞 LPD(n=4)。靶向测序显示,4 例患者存在 TET2-RHOAG17V 关联突变(如 AITL 中常见),另 1 例患者存在 TET2/DNMT3A/PLCG1/SETD2 突变谱。后一位患者,一位存在 TET2-RHOA 关联突变,另一位未检测到突变,出现了全身疾病并死亡。另外 5 例患者分别存在 TET2(n=1)、PLCG1(n=2)、SETD2(n=1)或 STAT5B(n=1)的孤立突变。
pcTFH-PTCL 患者具有与系统性 TFH 来源淋巴瘤重叠的病理和遗传特征。临床上,大多数患者仍局限于皮肤,只有 3 例患者出现全身扩散和死亡。pcTFH-PTCL 是否应在未来的分类中被纳入作为 TFH 淋巴瘤的一个新亚组,这仍然存在争议。关于这个话题,已知的有哪些?存在一组表达 T 滤泡辅助(TFH)标志物的皮肤淋巴瘤,它们似乎与现有的世界卫生组织诊断实体并不对应。这些包括蕈样真菌病、Sézary 综合征,或原发性皮肤 CD4 小/中 T 细胞淋巴增生性疾病,或伴有 TFH 表型的系统性外周 T 细胞淋巴瘤(PTCL)的皮肤扩展。这项研究增加了哪些新内容?这是首次对原发性皮肤外周 T 细胞淋巴瘤伴 T 滤泡辅助表型(pcTFH-PTCL)患者进行分子特征分析的大型原始系列研究。pcTFH-PTCL 可能是一组独立的皮肤淋巴瘤,具有与系统性 TFH 淋巴瘤重叠的临床病理和分子特征,如血管免疫母细胞性 T 细胞淋巴瘤,不属于已知的皮肤淋巴瘤诊断群体。这对患者的治疗和随访有影响;需要在进一步的研究中更好地阐明其临床行为,以调整患者的管理。