New York Genome Center, New York, NY, USA.
Department of Systems Biology, Columbia University, New York, NY, USA.
Nature. 2022 Aug;608(7922):353-359. doi: 10.1038/s41586-022-05035-y. Epub 2022 Aug 3.
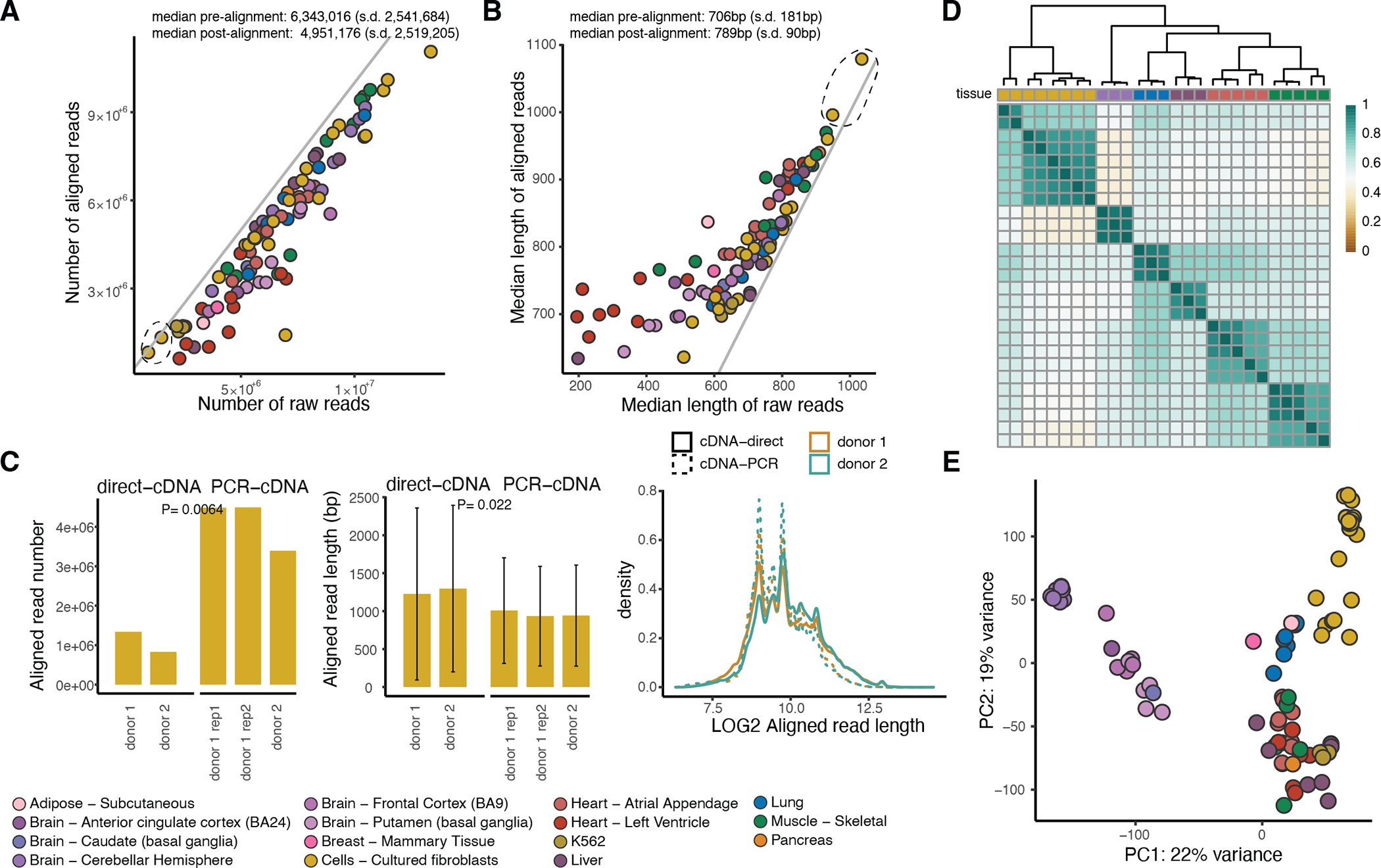
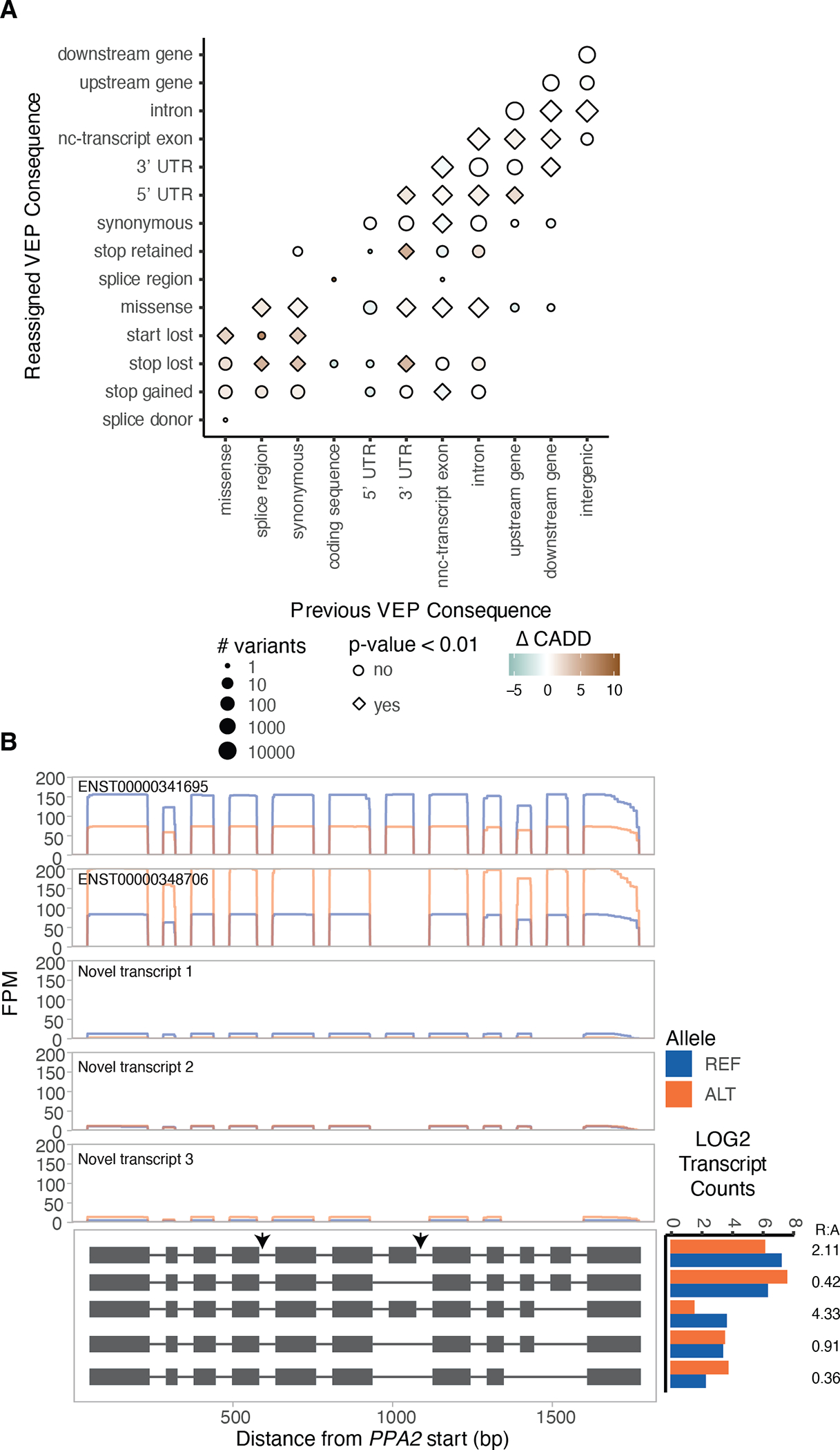
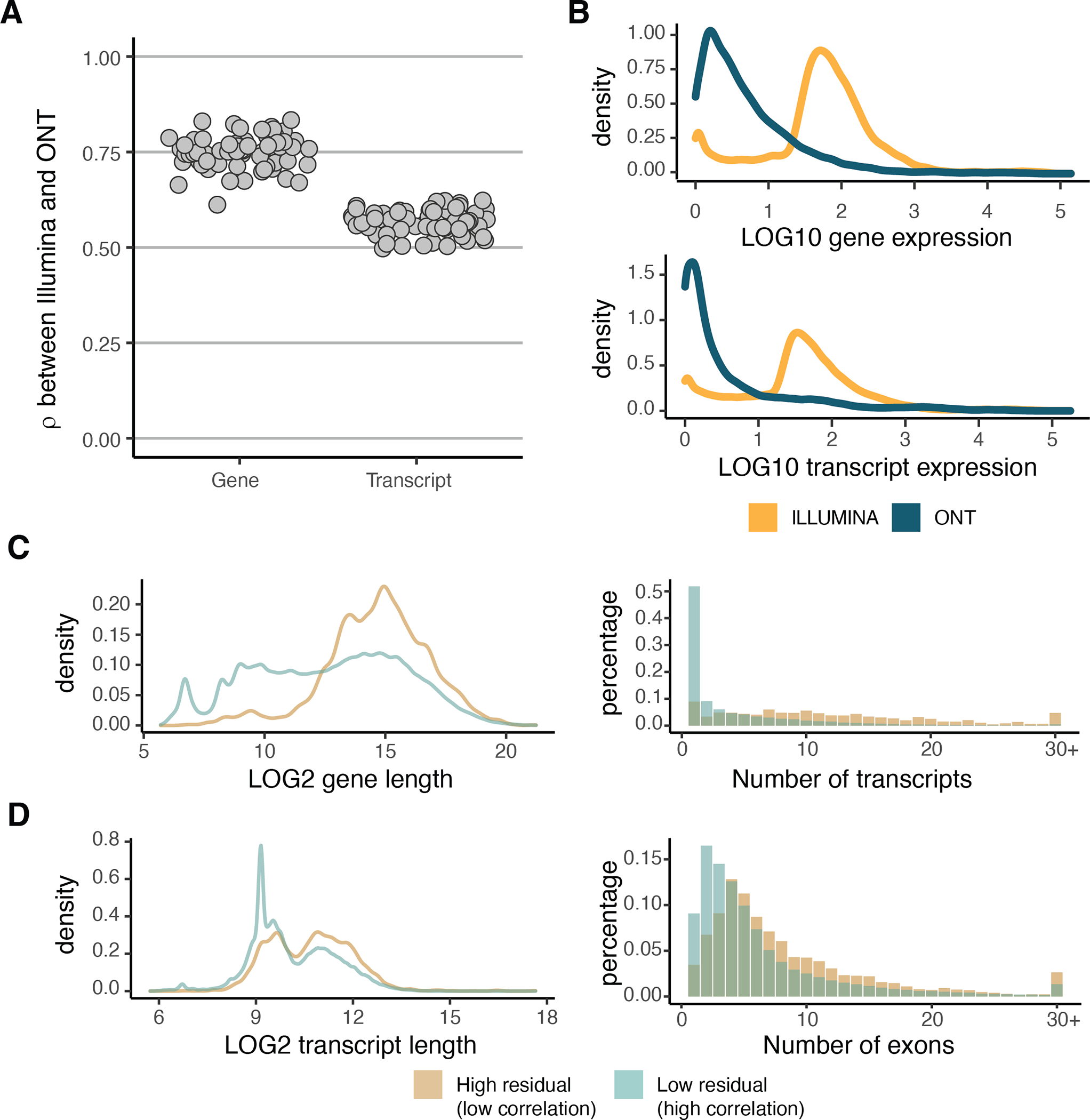
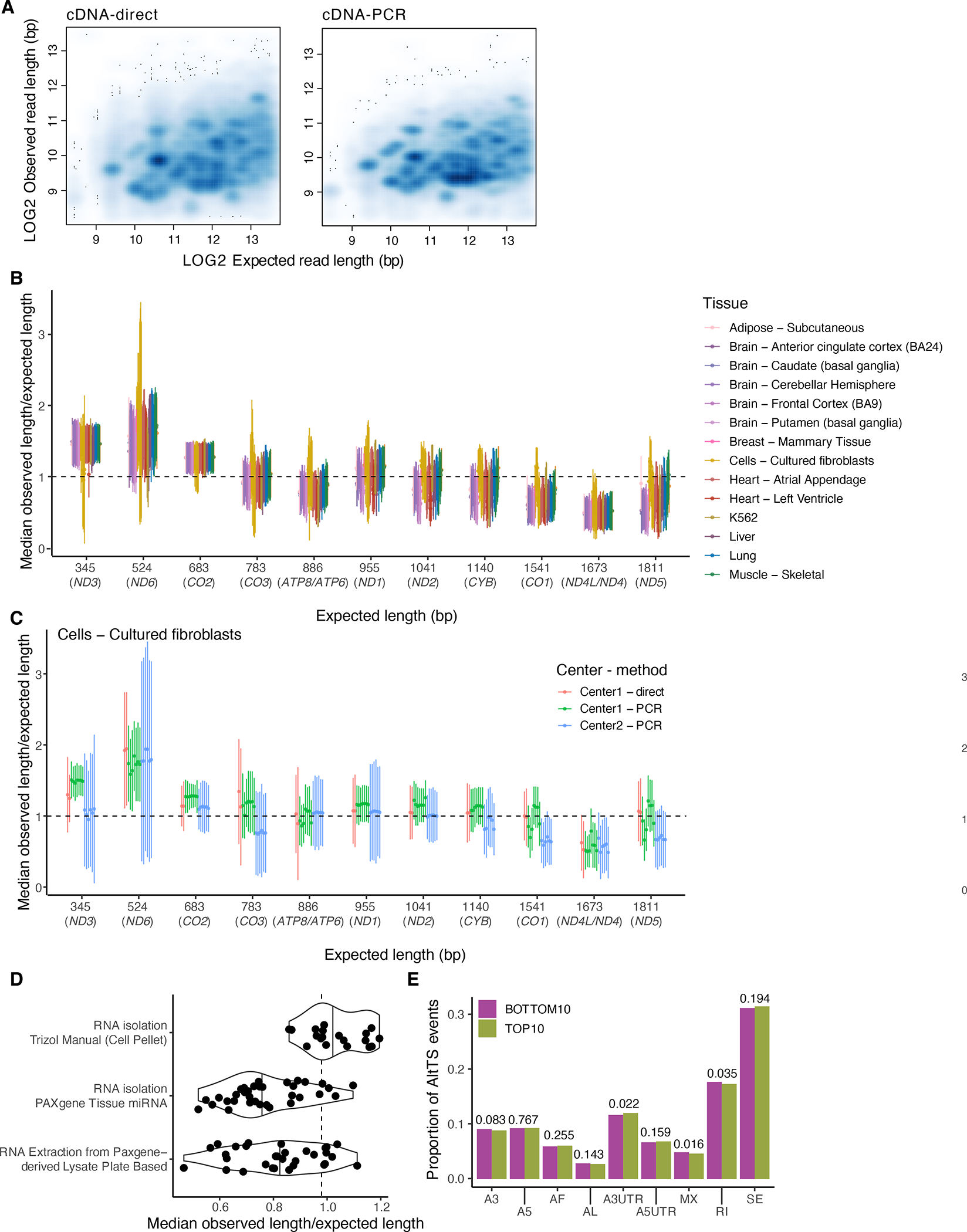
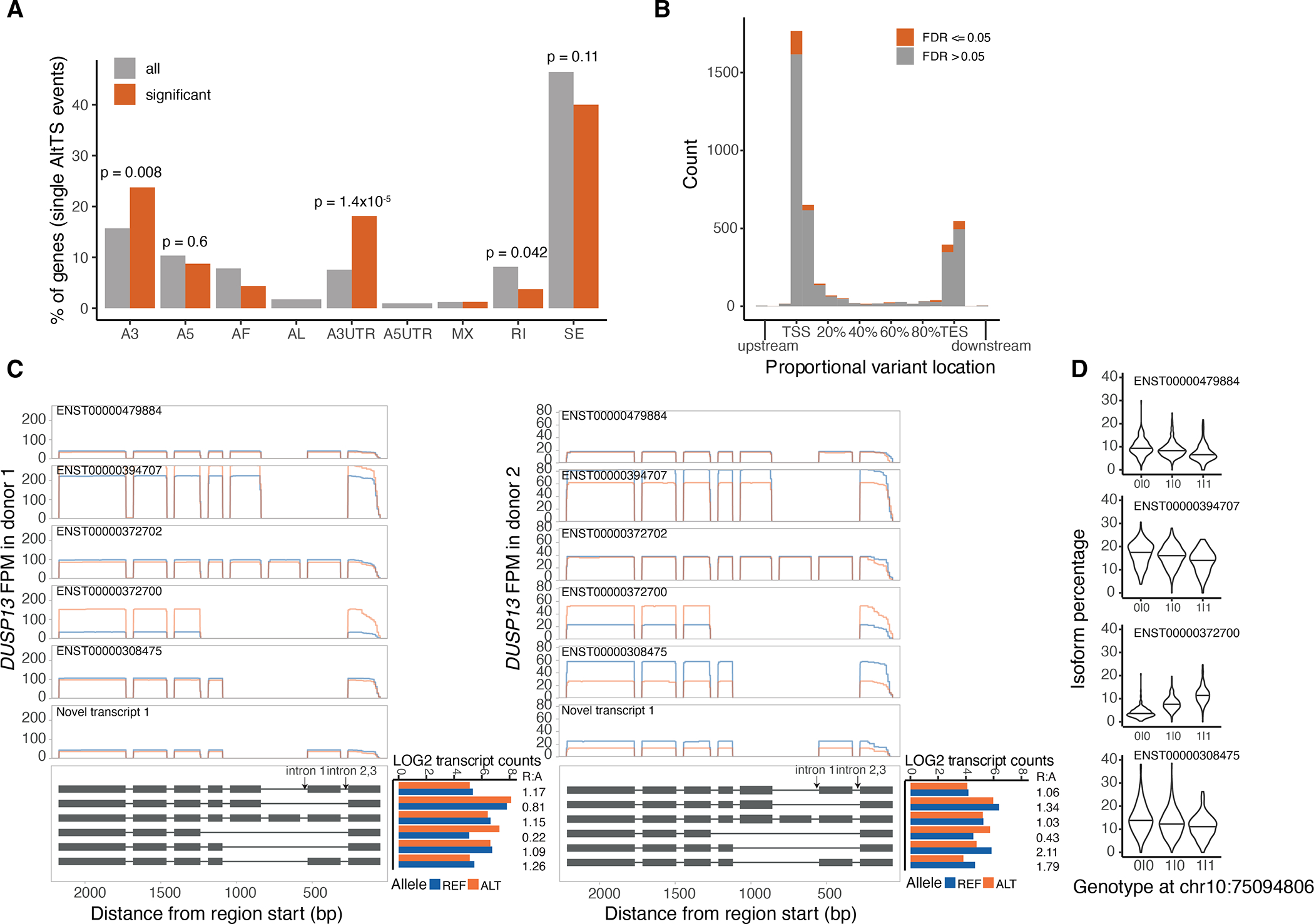
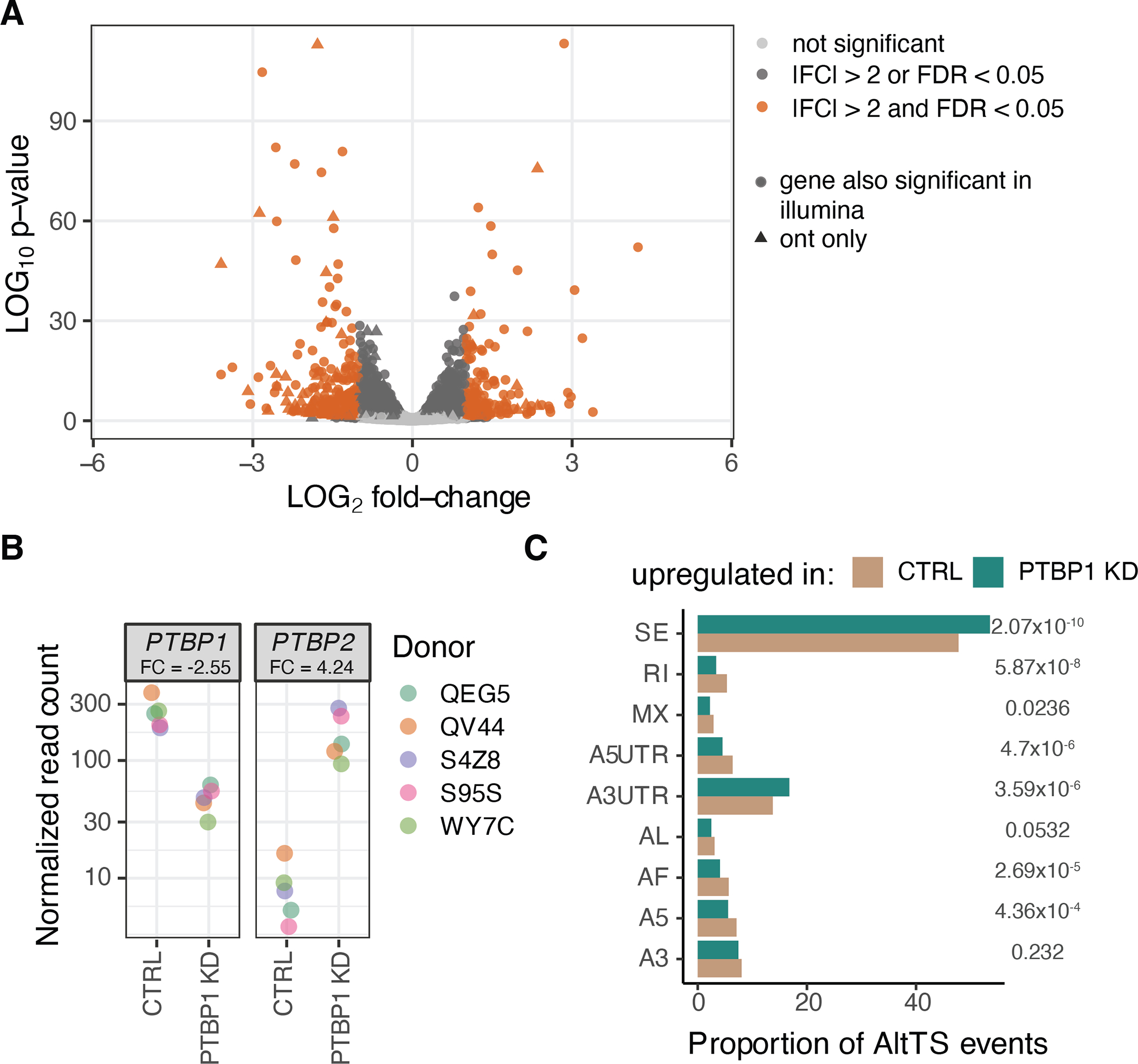
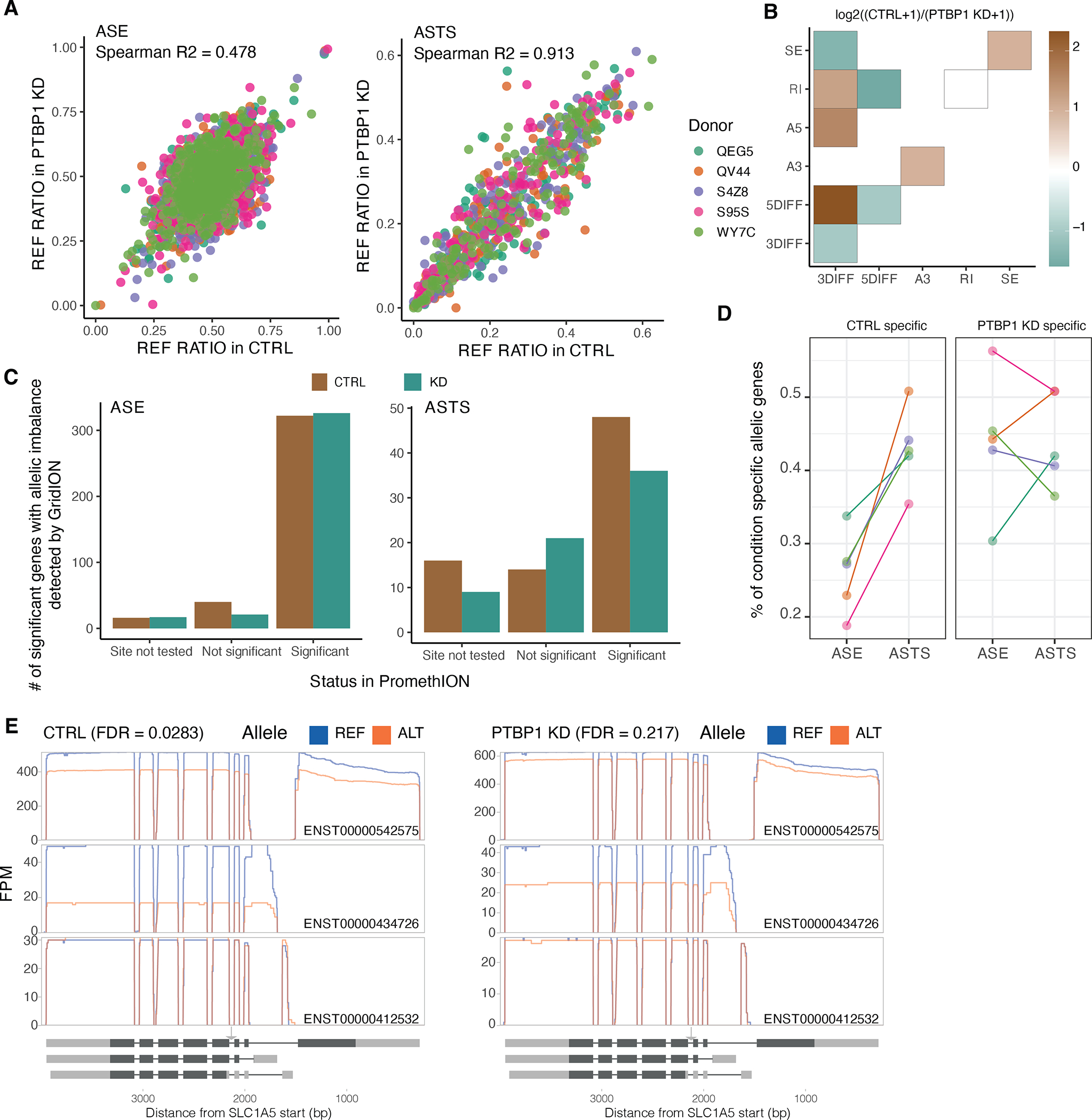
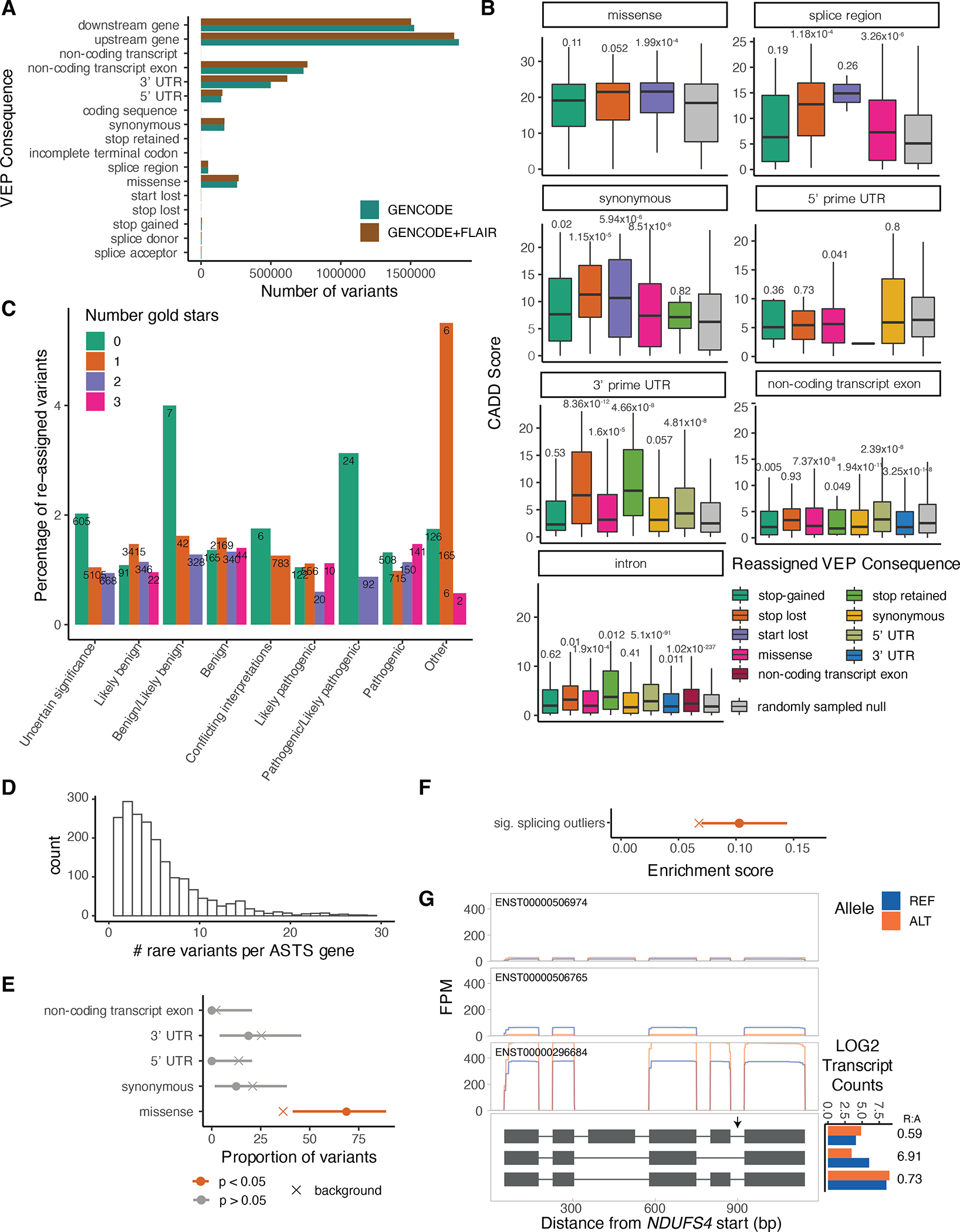
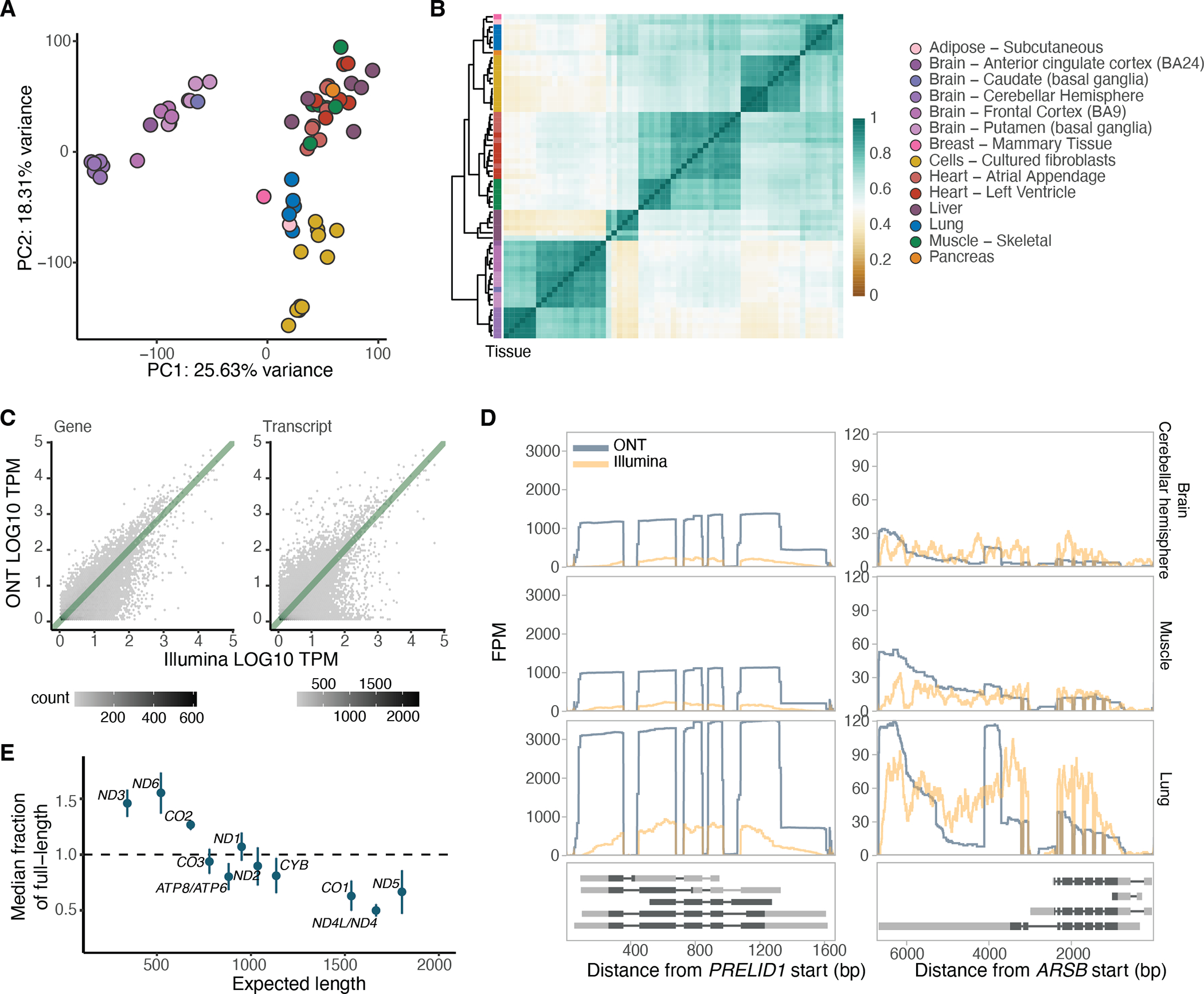
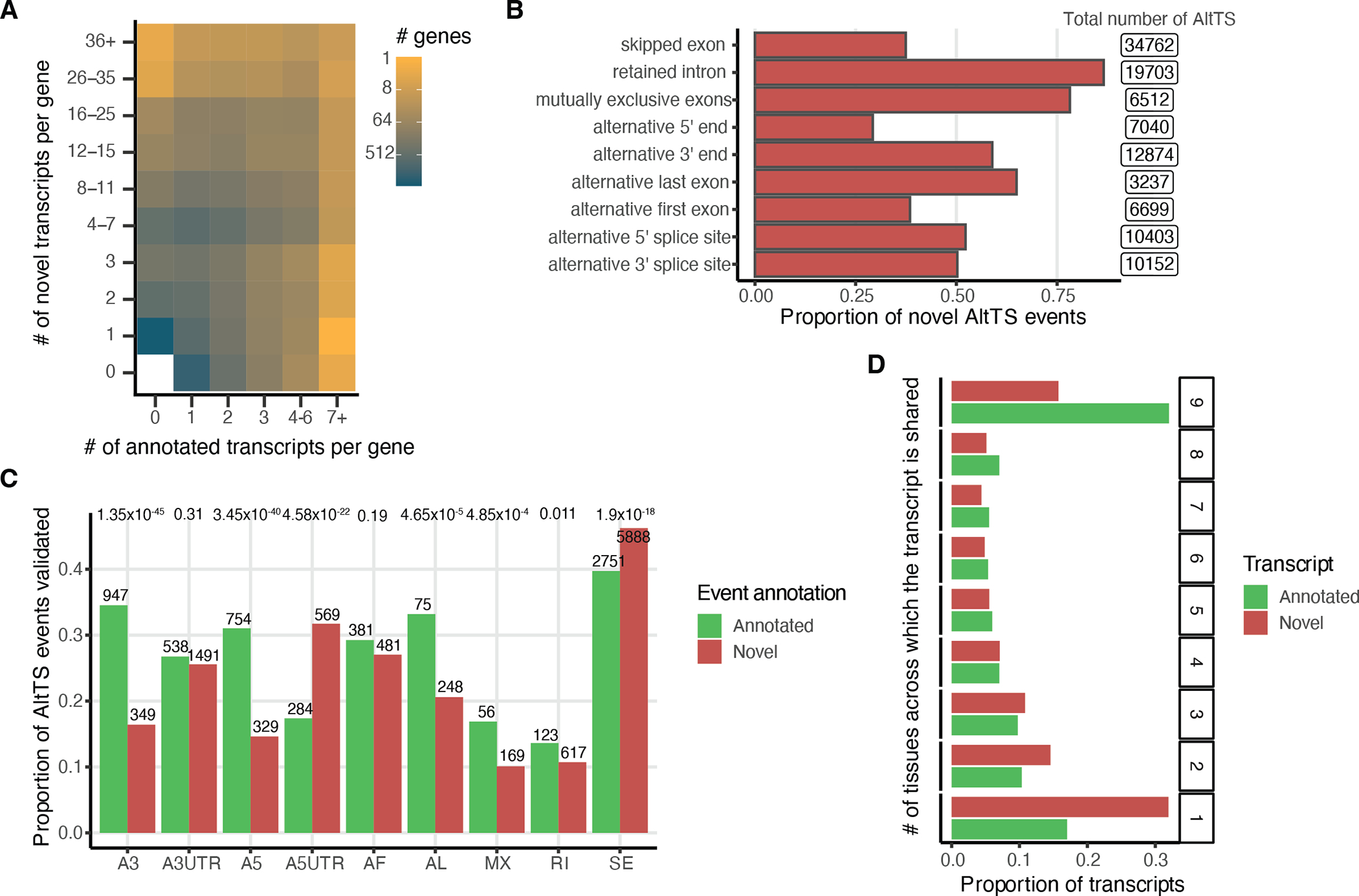
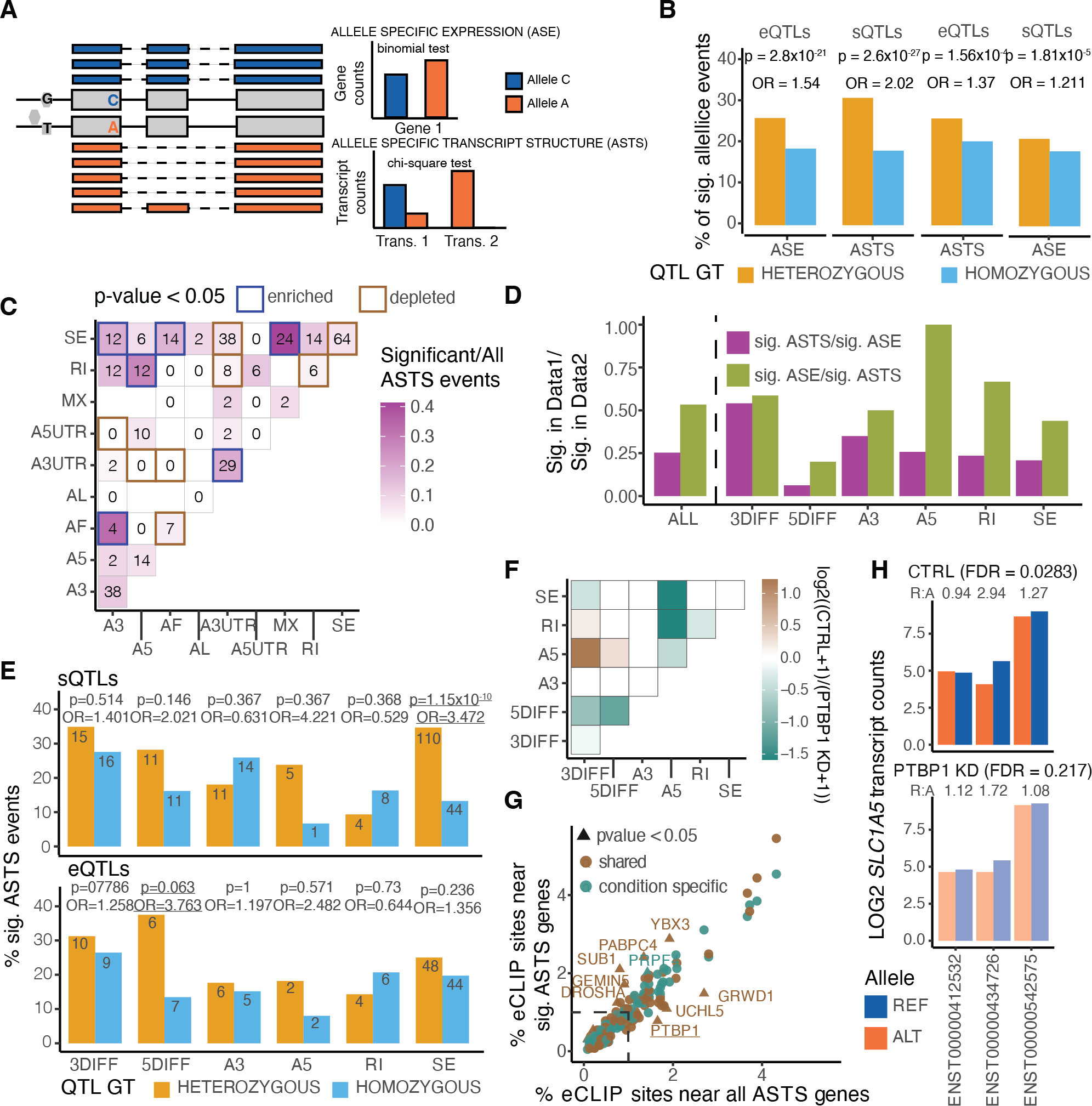
Regulation of transcript structure generates transcript diversity and plays an important role in human disease. The advent of long-read sequencing technologies offers the opportunity to study the role of genetic variation in transcript structure. In this Article, we present a large human long-read RNA-seq dataset using the Oxford Nanopore Technologies platform from 88 samples from Genotype-Tissue Expression (GTEx) tissues and cell lines, complementing the GTEx resource. We identified just over 70,000 novel transcripts for annotated genes, and validated the protein expression of 10% of novel transcripts. We developed a new computational package, LORALS, to analyse the genetic effects of rare and common variants on the transcriptome by allele-specific analysis of long reads. We characterized allele-specific expression and transcript structure events, providing new insights into the specific transcript alterations caused by common and rare genetic variants and highlighting the resolution gained from long-read data. We were able to perturb the transcript structure upon knockdown of PTBP1, an RNA binding protein that mediates splicing, thereby finding genetic regulatory effects that are modified by the cellular environment. Finally, we used this dataset to enhance variant interpretation and study rare variants leading to aberrant splicing patterns.
转录结构的调控产生转录本多样性,并在人类疾病中发挥重要作用。长读测序技术的出现为研究转录结构中遗传变异的作用提供了机会。在本文中,我们展示了一个来自 88 个基因型组织表达(GTEx)组织和细胞系样本的人类长读 RNA-seq 数据集,补充了 GTEx 资源。我们为注释基因鉴定了略多于 70000 个新的转录本,并验证了 10%新转录本的蛋白质表达。我们开发了一个新的计算软件包 LORALS,通过长读的等位基因特异性分析来分析稀有和常见变异对转录组的遗传效应。我们描述了等位基因特异性表达和转录本结构事件,为常见和稀有遗传变异引起的特定转录本改变提供了新的见解,并突出了长读数据带来的分辨率提高。我们能够通过敲低 RNA 结合蛋白 PTBP1 来干扰转录本结构,从而发现受细胞环境修饰的遗传调控效应。最后,我们使用这个数据集来增强变体解释,并研究导致异常剪接模式的罕见变体。