Lezcano María Ángeles, Sánchez-García Laura, Quesada Antonio, Carrizo Daniel, Fernández-Martínez Miguel Ángel, Cavalcante-Silva Erika, Parro Víctor
Centro de Astrobiología (CAB), CSIC-INTA, Carretera de Ajalvir, Madrid, Spain.
Departamento de Biología, C. Darwin 2, Universidad Autónoma de Madrid, Madrid, Spain.
Front Microbiol. 2022 Apr 12;13:799360. doi: 10.3389/fmicb.2022.799360. eCollection 2022.
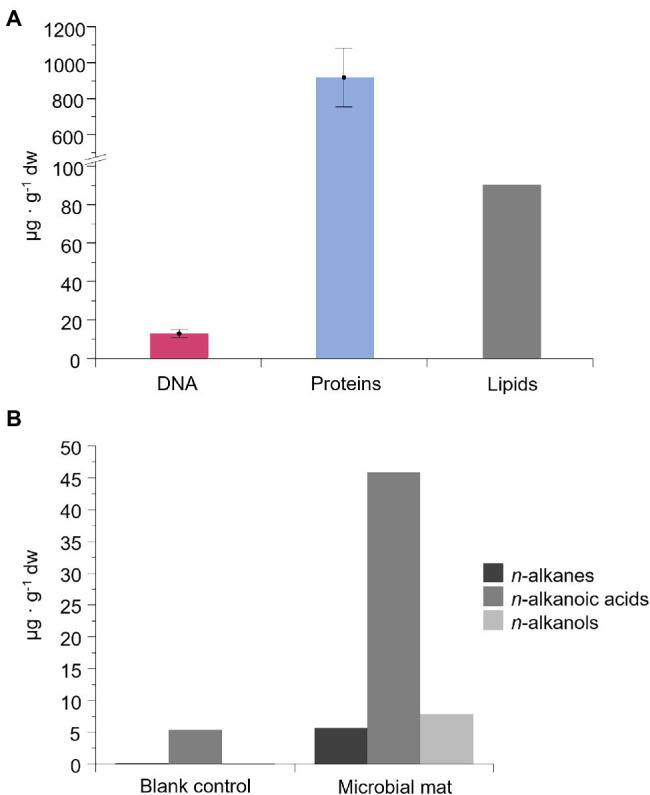
Paleobiological reconstructions based on molecular fossils may be limited by degradation processes causing differential preservation of biomolecules, the distinct taxonomic specificity of each biomolecule type, and analytical biases. Here, we combined the analysis of DNA, proteins and lipid biomarkers using 16S and 18S rRNA gene metabarcoding, metaproteomics and lipid analysis to reconstruct the taxonomic composition and metabolisms of a desiccated microbial mat from the McMurdo Ice Shelf (MIS) (Antarctica) dated ~1,000 years BP. The different lability, taxonomic resolution and analytical bias of each biomolecule type led to a distinct microbial community profile. DNA analysis showed selective preservation of DNA remnants from the most resistant taxa (e.g., spore-formers). In contrast, the proteins profile revealed microorganisms missed by DNA sequencing, such as , and showed a microbial composition similar to fresh microbial mats in the MIS. Lipid hydrocarbons also confirmed and suggested the presence of mosses or vascular plant remnants from a period in Antarctica when the climate was warmer (e.g., Mid-Miocene or Eocene). The combined analysis of the three biomolecule types also revealed diverse metabolisms that operated in the microbial mat before desiccation: oxygenic and anoxygenic photosynthesis, nitrogen fixation, nitrification, denitrification, sulfur reduction and oxidation, and methanogenesis. Therefore, the joint analysis of DNA, proteins and lipids resulted in a powerful approach that improved taxonomic and metabolic reconstructions overcoming information gaps derived from using individual biomolecules types.
基于分子化石的古生物学重建可能受到多种因素的限制,包括导致生物分子差异保存的降解过程、每种生物分子类型独特的分类特异性以及分析偏差。在这里,我们结合了DNA、蛋白质和脂质生物标志物的分析,使用16S和18S rRNA基因宏条形码、宏蛋白质组学和脂质分析,来重建来自南极麦克默多冰架(MIS)的一块距今约1000年的干化微生物垫的分类组成和代谢情况。每种生物分子类型不同的稳定性、分类分辨率和分析偏差导致了独特的微生物群落概况。DNA分析显示,来自最具抗性的分类群(如产孢菌)的DNA残余物被选择性保存。相比之下,蛋白质概况揭示了DNA测序遗漏的微生物,如,并显示出与MIS中新鲜微生物垫相似的微生物组成。脂质碳氢化合物也证实了这一点,并表明在南极气候较温暖的时期(如中新世中期或始新世)存在苔藓或维管植物残余物。对这三种生物分子类型的综合分析还揭示了微生物垫在干燥之前所进行的多种代谢:有氧和无氧光合作用、固氮、硝化作用、反硝化作用、硫还原和氧化以及甲烷生成。因此,对DNA、蛋白质和脂质的联合分析产生了一种强大的方法,该方法改进了分类和代谢重建,克服了使用单一生物分子类型所产生的信息缺口。