Center for Molecular Medicine and Genetics, Detroit, MI 48201, USA.
Departments of Ophthalmology Visual Anatomical Science and pharmacology, Wayne State University School of Medicine, Detroit, MI 48201, USA.
Mol Metab. 2022 Oct;64:101562. doi: 10.1016/j.molmet.2022.101562. Epub 2022 Aug 6.
The mitochondrial nicotinamide adenine dinucleotide (NAD) kinase (MNADK) mediates de novo mitochondrial NADP biosynthesis by catalyzing the phosphorylation of NAD to yield NADP. In this study, we investigated the function and mechanistic basis by which MNADK regulates metabolic homeostasis.
Generalized gene set analysis by aggregating human patient genomic databases, metabolic studies with genetically engineered animal models, mitochondrial bioenergetic analysis, as well as gain- and loss- of-function studies were performed to address the functions and mechanistic basis by which MNADK regulates energy metabolism and redox state associated with metabolic disease.
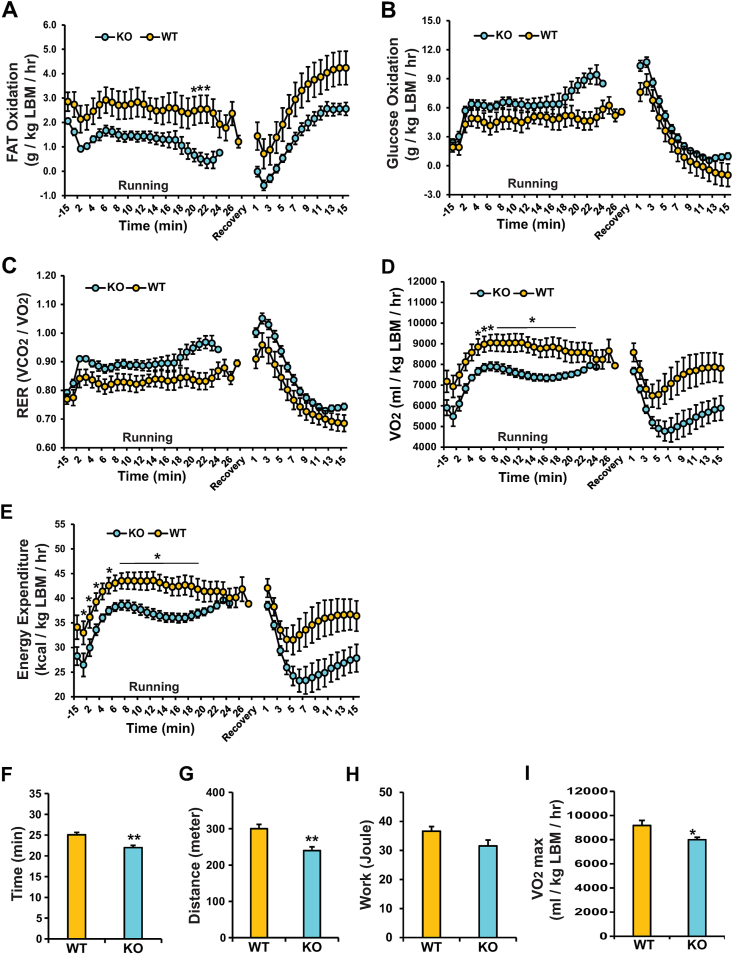
Human MNADK common gene variants or decreased expression of the gene are significantly associated with the occurrence of type-2 diabetes, non-alcoholic fatty liver disease (NAFLD), or hepatocellular carcinoma (HCC). Ablation of the MNADK gene in mice led to decreased fat oxidation, coincident with increased respiratory exchange ratio (RER) and decreased energy expenditure upon energy demand triggered by endurance exercise or fasting. On an atherogenic high-fat diet (HFD), MNADK-null mice exhibited hepatic insulin resistance and glucose intolerance, indicating a type-2 diabetes-like phenotype in the absence of MNADK. MNADK deficiency led to a decrease in mitochondrial NADP(H) but an increase in cellular reactive oxygen species (ROS) in mouse livers. Consistently, protein levels of the major metabolic regulators or enzymes were decreased, while their acetylation modifications were increased in the livers of MNADK-null mice. Feeding mice with a HFD caused S-nitrosylation (SNO) modification, a posttranslational modification that represses protein activities, on MNADK protein in the liver. Reconstitution of an SNO-resistant MNADK variant, MNADK-S193, into MNADK-null mice mitigated hepatic steatosis induced by HFD.
MNADK, the only known mammalian mitochondrial NAD kinase, plays important roles in preserving energy homeostasis to mitigate the risk of metabolic disorders.
线粒体烟酰胺腺嘌呤二核苷酸(NAD)激酶(MNADK)通过催化 NAD 的磷酸化生成 NADP,介导从头合成线粒体 NADP。在这项研究中,我们研究了 MNADK 调节代谢平衡的功能和机制基础。
通过聚合人类患者基因组数据库进行广义基因集分析、利用基因工程动物模型进行代谢研究、线粒体生物能分析以及获得和丧失功能研究,来解决 MNADK 调节与代谢疾病相关的能量代谢和氧化还原状态的功能和机制基础。
人类 MNADK 常见基因变异或基因表达降低与 2 型糖尿病、非酒精性脂肪性肝病(NAFLD)或肝细胞癌(HCC)的发生显著相关。在小鼠中敲除 MNADK 基因导致脂肪氧化减少,同时伴随着耐力运动或禁食触发的能量需求时呼吸交换率(RER)增加和能量消耗减少。在动脉粥样硬化高脂饮食(HFD)中,MNADK 缺失的小鼠表现出肝胰岛素抵抗和葡萄糖不耐受,表明在没有 MNADK 的情况下出现 2 型糖尿病样表型。MNADK 缺乏导致肝线粒体 NADP(H)减少,但细胞内活性氧(ROS)增加。一致地,MNADK 缺失小鼠肝脏中的主要代谢调节剂或酶的蛋白水平降低,但其乙酰化修饰增加。高脂饮食喂养导致 MNADK 蛋白在肝脏中的 S-亚硝基化(SNO)修饰,SNO 是一种抑制蛋白质活性的翻译后修饰。将 SNO 抗性 MNADK 变体 MNADK-S193 重建到 MNADK 缺失的小鼠中,可减轻 HFD 引起的肝脂肪变性。
MNADK 是唯一已知的哺乳动物线粒体 NAD 激酶,在维持能量平衡方面发挥着重要作用,以降低代谢紊乱的风险。