Department of Microbiology and Immunology, The University of Western Ontario, London, Ontario, Canada.
Department of Pathology and Laboratory Medicine, The University of Western Ontario, London, Ontario, Canada.
mSphere. 2022 Aug 31;7(4):e0031722. doi: 10.1128/msphere.00317-22. Epub 2022 Aug 11.
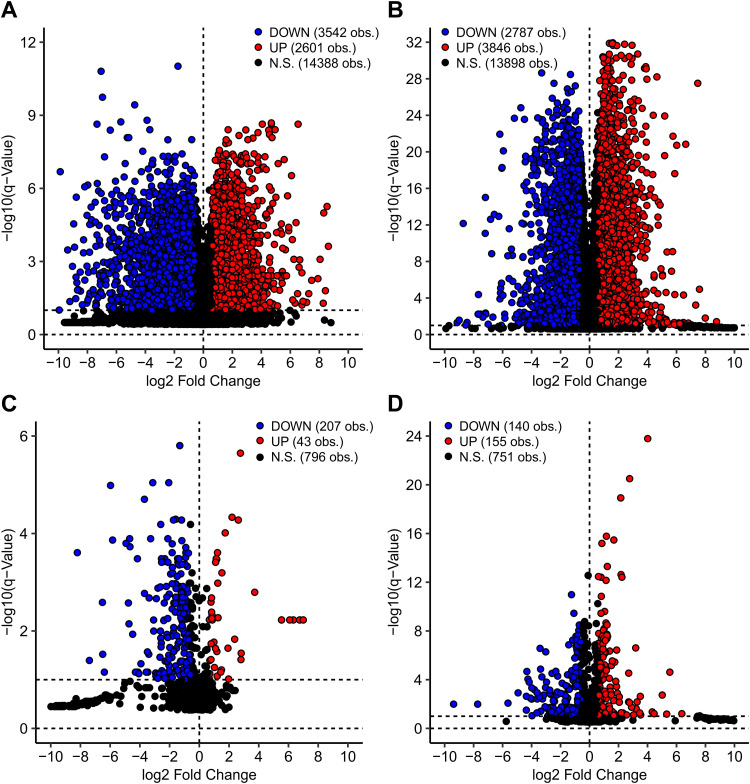
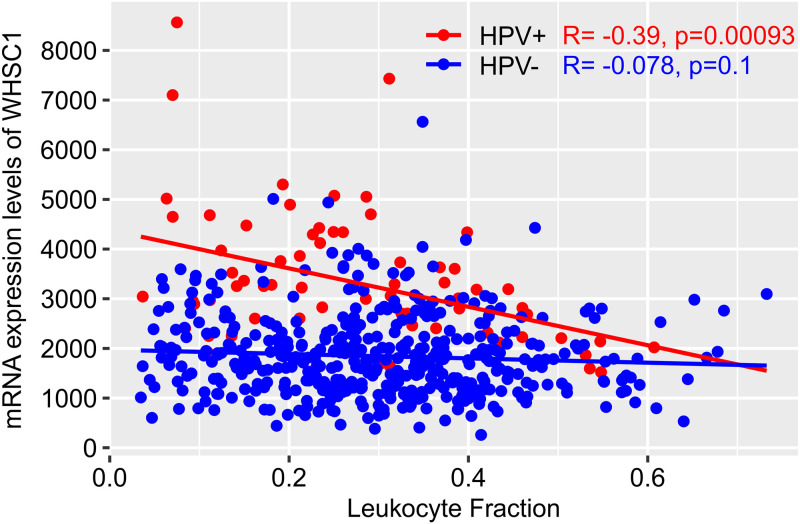
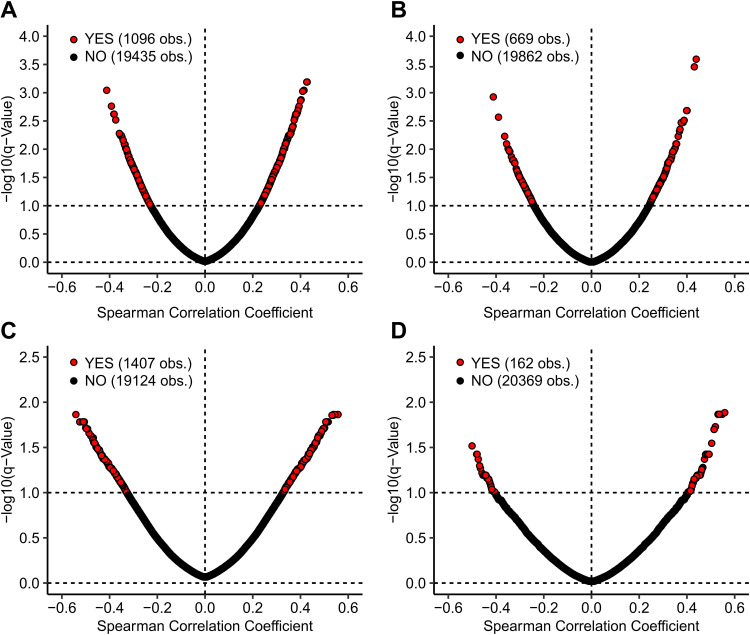
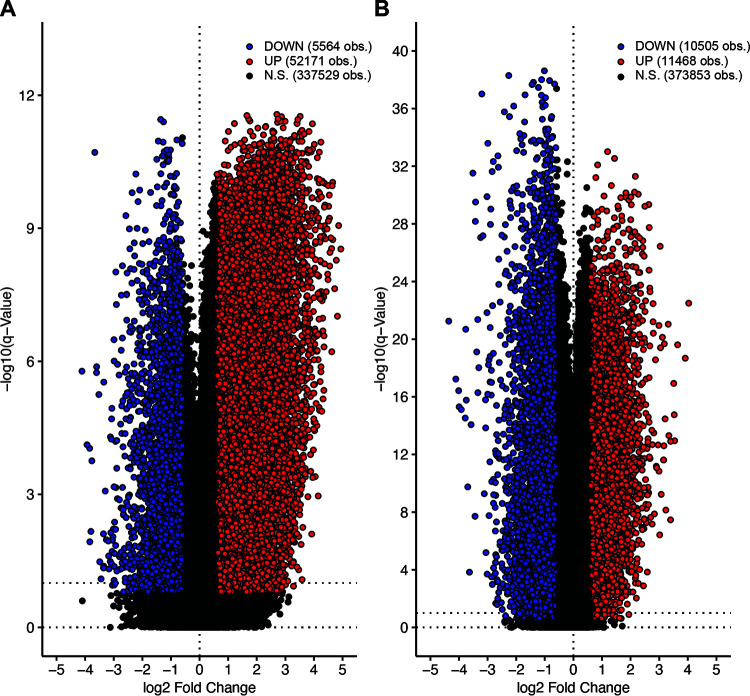
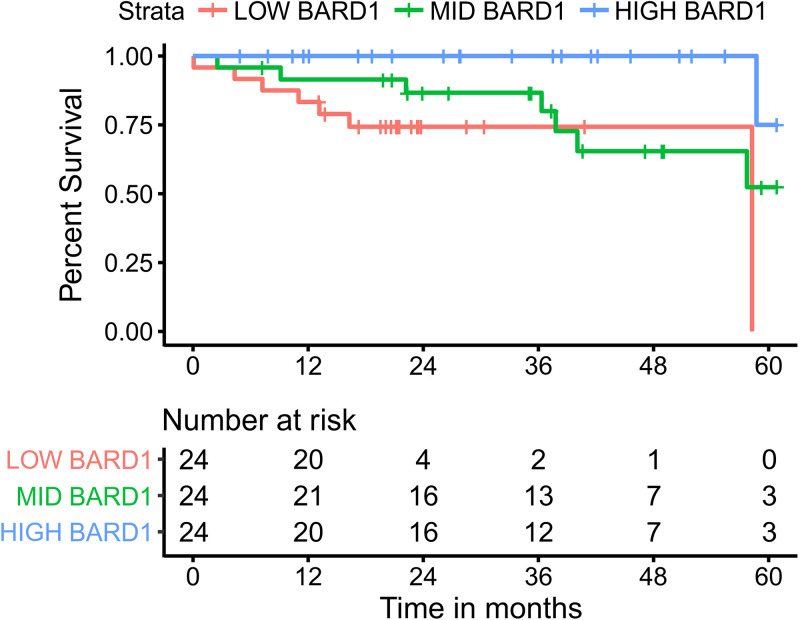
Human papillomaviruses (HPVs) are highly infectious and cause the most common sexually transmitted viral infections. They induce hyperproliferation of squamous epithelial tissue, often forming warts. Virally encoded proteins reprogram gene expression and cell growth to create an optimal environment for viral replication. In addition to their normal roles in infection, functional alterations induced by viral proteins establish conditions that frequently contribute to human carcinogenesis. In fact, ~5% of human cancers are caused by HPVs, with virtually all cervical squamous cell carcinomas (CESC) and an increasing number of head and neck squamous cell carcinomas (HNSC) attributed to HPV infection. The Cancer Genome Atlas (TCGA) molecularly characterized thousands of primary human cancer samples in many cancer types, including CESC and HNSC, and created a comprehensive atlas of genomic, epigenomic, and transcriptomic data. This publicly available genome-wide information provides an unprecedented opportunity to expand the knowledge of the role that HPV plays in human carcinogenesis. While many tools exist to mine these data, few, if any, focus on the comparison of HPV-positive cancers with their HPV-negative counterparts or adjacent normal control tissue. We have constructed a suite of web-based tools, The HPV Induced Cancer Resource (THInCR), to utilize TCGA data for research related to HPV-induced CESC and HNSC. These tools allow investigators to gain greater biological and medical insights by exploring the impacts of HPV on cellular gene expression (mRNA and microRNA), altered gene methylation, and associations with patient survival and immune landscape features. These tools are accessible at https://thincr.ca/. The suite of analytical tools of THInCR provides the opportunity to investigate the roles that candidate target genes identified in cell lines or other model systems contribute to in actual HPV-dependent human cancers and is based on large-scale TCGA data sets. Expression of target genes, including both mRNA and microRNA, can be correlated with HPV gene expression, epigenetic changes in DNA methylation, patient survival, and numerous immune features, like leukocyte infiltration, interferon gamma response, T cell response, etc. Data from these analyses may immediately provide evidence to validate observations, reveal insights into mechanisms of virus-mediated alterations in cell growth, behavior, gene expression, and innate and adaptive immunity and may help hypothesis generation for further investigations.
人乳头瘤病毒(HPV)具有高度传染性,可引起最常见的性传播病毒感染。它们诱导鳞状上皮组织的过度增殖,常形成疣。病毒编码的蛋白重新编程基因表达和细胞生长,为病毒复制创造最佳环境。除了在感染中的正常作用外,病毒蛋白引起的功能改变建立了经常促进人类癌变的条件。事实上,约 5%的人类癌症是由 HPV 引起的,几乎所有的宫颈鳞状细胞癌(CESC)和越来越多的头颈部鳞状细胞癌(HNSC)都归因于 HPV 感染。癌症基因组图谱(TCGA)对包括 CESC 和 HNSC 在内的许多癌症类型的数千个人类原发性癌症样本进行了分子特征分析,并创建了基因组、表观基因组和转录组数据的综合图谱。这些公开的全基因组信息提供了一个前所未有的机会,以扩展 HPV 在人类癌变中的作用的知识。虽然有许多工具可以挖掘这些数据,但很少有(如果有的话)工具专门用于比较 HPV 阳性癌症与其 HPV 阴性对应物或相邻正常对照组织。我们构建了一套基于网络的工具,即 HPV 诱导性癌症资源(THInCR),用于利用 TCGA 数据进行与 HPV 诱导的 CESC 和 HNSC 相关的研究。这些工具允许研究人员通过探索 HPV 对细胞基因表达(mRNA 和 microRNA)、基因甲基化改变以及与患者生存和免疫景观特征的关联,获得更大的生物学和医学见解。这些工具可在 https://thincr.ca/ 上获得。THInCR 的分析工具套件提供了一个机会,可以研究在细胞系或其他模型系统中鉴定的候选靶基因在实际 HPV 依赖性人类癌症中所起的作用,这些研究是基于大规模的 TCGA 数据集进行的。靶基因的表达,包括 mRNA 和 microRNA,可以与 HPV 基因表达、DNA 甲基化的表观遗传改变、患者生存以及白细胞浸润、干扰素γ反应、T 细胞反应等众多免疫特征相关联。这些分析的数据可能立即提供证据来验证观察结果,揭示病毒介导的细胞生长、行为、基因表达以及先天和适应性免疫改变的机制中的见解,并可能有助于为进一步的研究生成假说。