Center for Integrative Genomics, University of Lausanne, Lausanne, Switzerland.
Swiss Institute of Bioinformatics, Lausanne, Switzerland.
Genome Med. 2022 Aug 11;14(1):89. doi: 10.1186/s13073-022-01088-w.
The genetic underpinning of sexual dimorphism is very poorly understood. The prevalence of many diseases differs between men and women, which could be in part caused by sex-specific genetic effects. Nevertheless, only a few published genome-wide association studies (GWAS) were performed separately in each sex. The reported enrichment of expression quantitative trait loci (eQTLs) among GWAS-associated SNPs suggests a potential role of sex-specific eQTLs in the sex-specific genetic mechanism underlying complex traits.
To explore this scenario, we combined sex-specific whole blood RNA-seq eQTL data from 3447 European individuals included in BIOS Consortium and GWAS data from UK Biobank. Next, to test the presence of sex-biased causal effect of gene expression on complex traits, we performed sex-specific transcriptome-wide Mendelian randomization (TWMR) analyses on the two most sexually dimorphic traits, waist-to-hip ratio (WHR) and testosterone levels. Finally, we performed power analysis to calculate the GWAS sample size needed to observe sex-specific trait associations driven by sex-biased eQTLs.
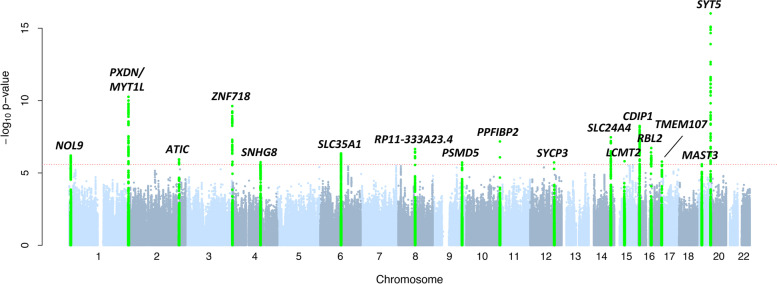
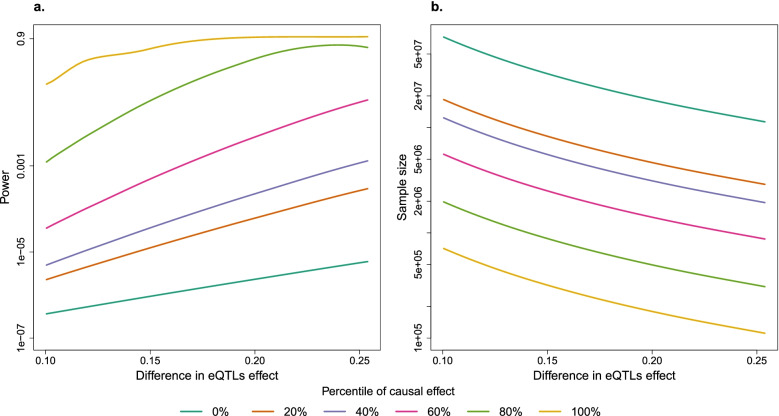
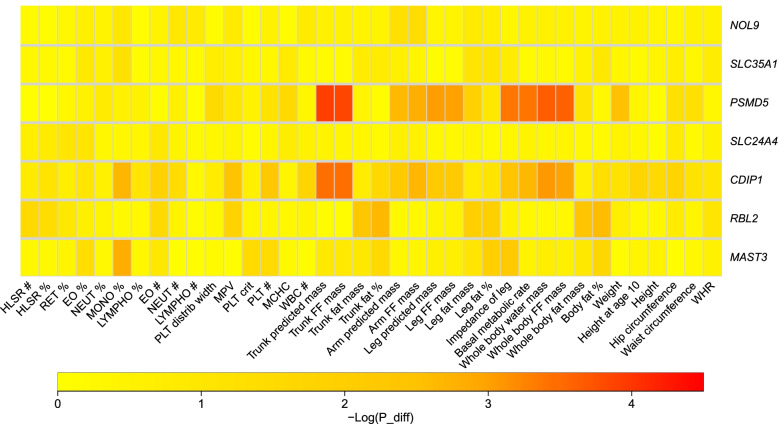
Among 9 million SNP-gene pairs showing sex-combined associations, we found 18 genes with significant sex-biased cis-eQTLs (FDR 5%). Our phenome-wide association study of the 18 top sex-biased eQTLs on >700 traits unraveled that these eQTLs do not systematically translate into detectable sex-biased trait-associations. In addition, we observed that sex-specific causal effects of gene expression on complex traits are not driven by sex-specific eQTLs. Power analyses using real eQTL- and causal-effect sizes showed that millions of samples would be necessary to observe sex-biased trait associations that are fully driven by sex-biased cis-eQTLs. Compensatory effects may further hamper their detection.
Our results suggest that sex-specific eQTLs in whole blood do not translate to detectable sex-specific trait associations of complex diseases, and vice versa that the observed sex-specific trait associations cannot be explained by sex-specific eQTLs.
性二态性的遗传基础还知之甚少。许多疾病在男性和女性中的发病率不同,这在一定程度上可能是由于性别特异性遗传效应所致。然而,之前只进行了少数几项分别在男性和女性中进行的全基因组关联研究(GWAS)。GWAS 相关 SNP 中表达数量性状基因座(eQTL)的富集表明,性别特异性 eQTL 在复杂性状的性别特异性遗传机制中可能发挥作用。
为了探索这种情况,我们结合了 BIOS 联盟中包含的 3447 名欧洲个体的全血 RNA-seq eQTL 数据和英国生物库的 GWAS 数据。接下来,为了测试基因表达对复杂性状的因果效应是否存在性别偏向,我们对两个最具性别二态性的性状(腰臀比(WHR)和睾丸酮水平)进行了性别特异性全转录组孟德尔随机化(TWMR)分析。最后,我们进行了功效分析,以计算观察由性别偏向 eQTL 驱动的性别特异性性状关联所需的 GWAS 样本量。
在表现出联合性别关联的 900 万个 SNP-基因对中,我们发现了 18 个具有显著性别偏向 cis-eQTL 的基因(FDR 为 5%)。我们对 >700 个性状的 18 个 top 性别偏向 eQTL 进行了全表型关联研究,结果表明这些 eQTL 不会系统地转化为可检测到的性别偏向性状关联。此外,我们观察到,基因表达对复杂性状的因果效应不是由性别特异性 eQTL 驱动的。使用真实的 eQTL 和因果效应大小进行的功效分析表明,需要数百万个样本才能观察到完全由性别偏向 cis-eQTL 驱动的性别偏向性状关联。补偿效应可能会进一步阻碍它们的检测。
我们的研究结果表明,全血中的性别特异性 eQTL 不会转化为复杂疾病的可检测的性别特异性性状关联,反之亦然,观察到的性别特异性性状关联不能用性别特异性 eQTL 来解释。