Department of Therapeutic Chemistry, National Research Center, Dokki, Cairo 12622, Egypt.
Institute for Computational Molecular Science, Department of Chemistry, Temple University, Philadelphia, PA 19122, USA.
Molecules. 2022 Aug 2;27(15):4924. doi: 10.3390/molecules27154924.
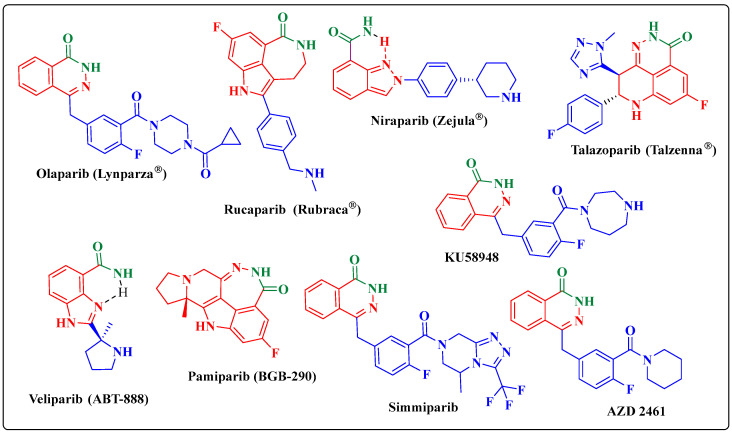
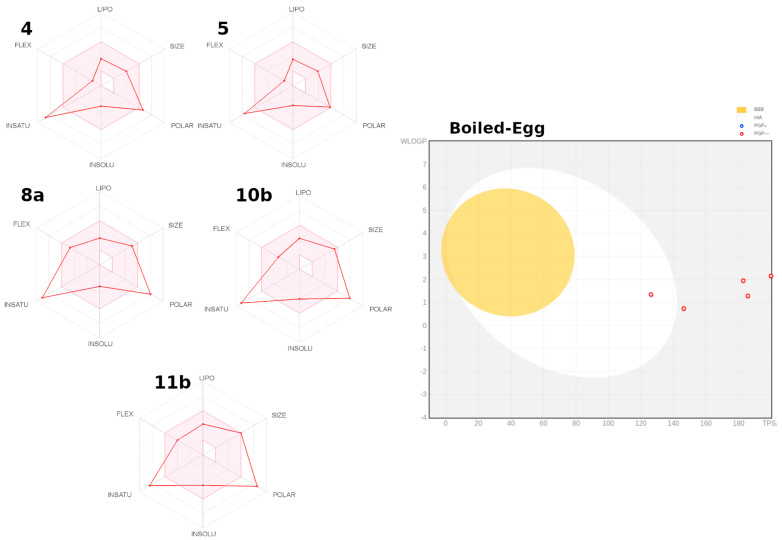
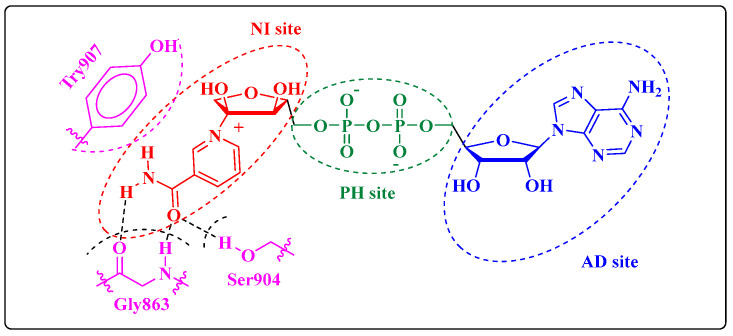
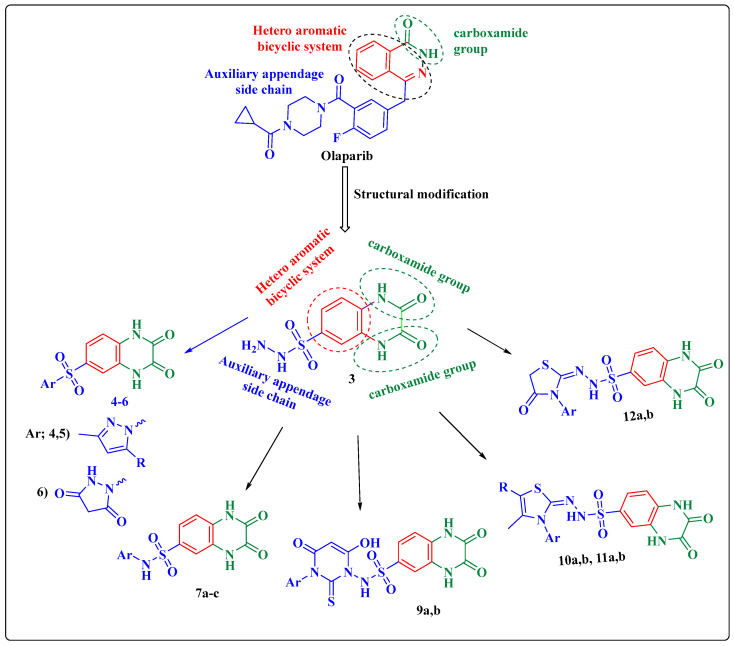
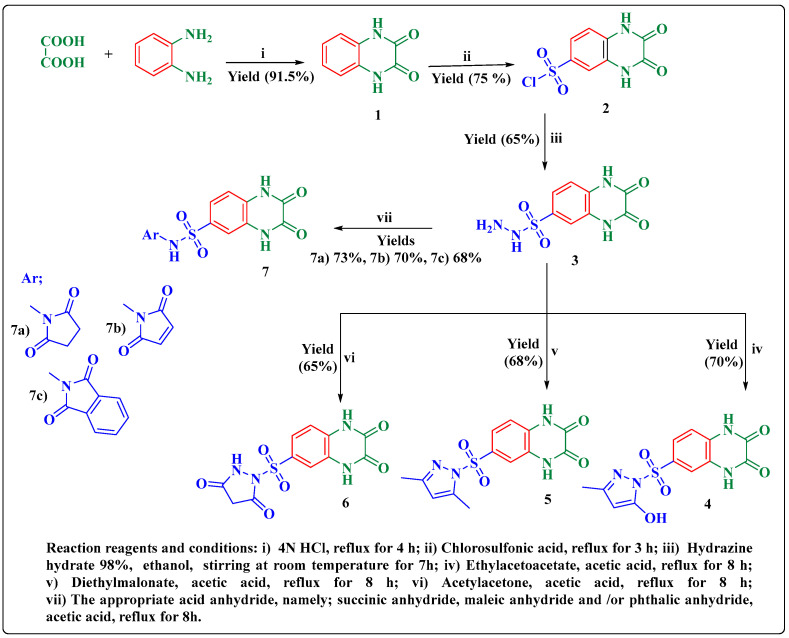
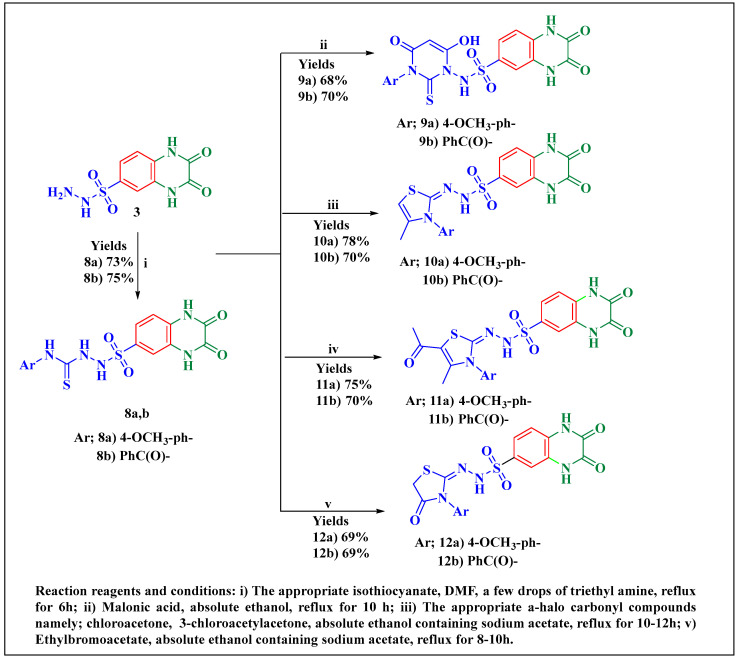
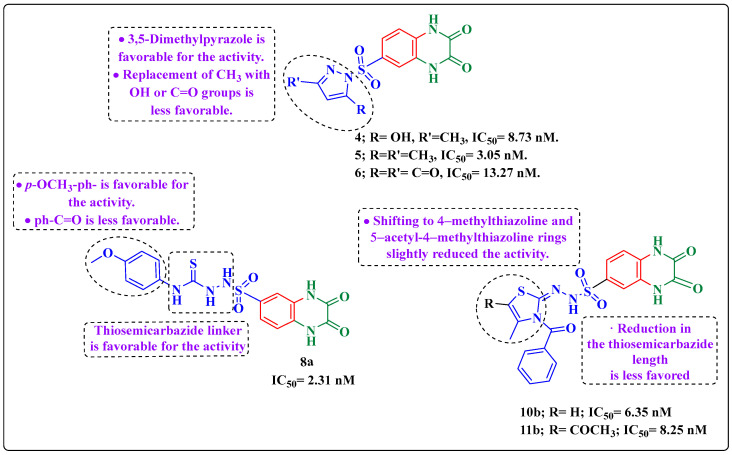
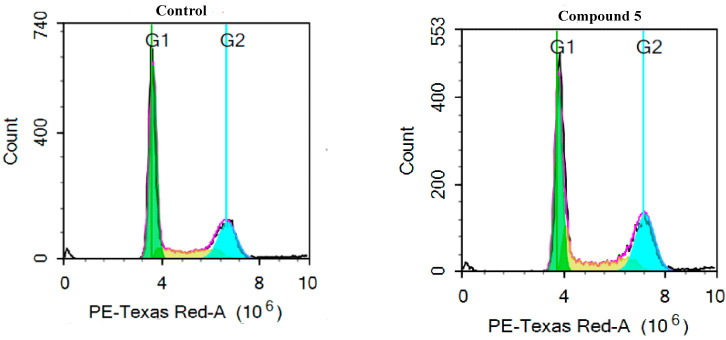
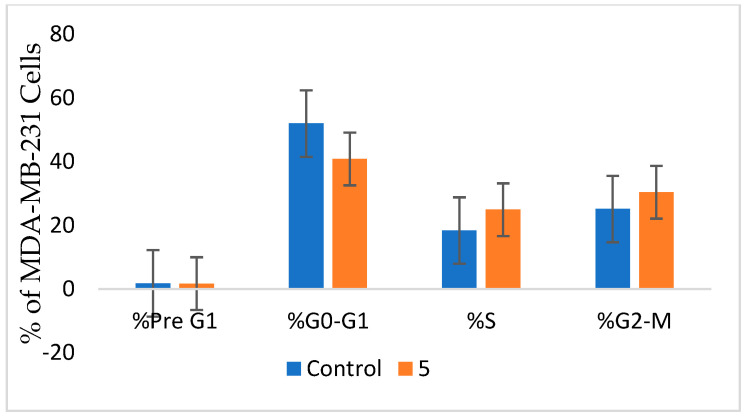
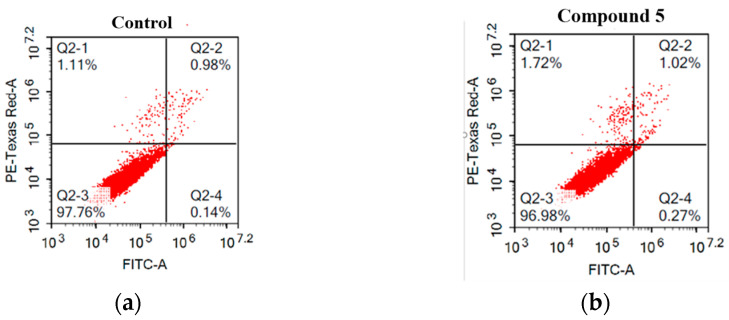
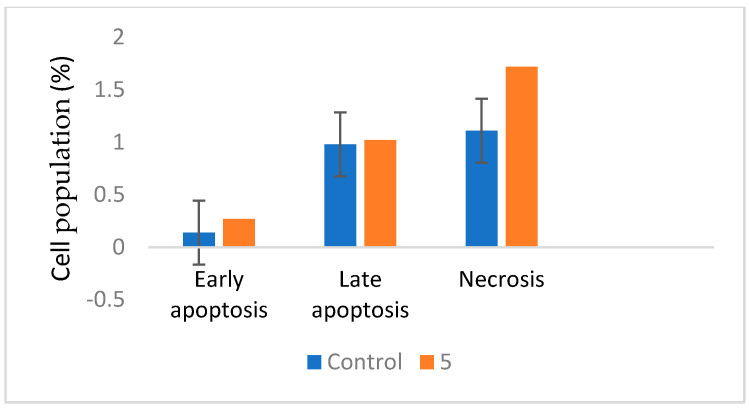
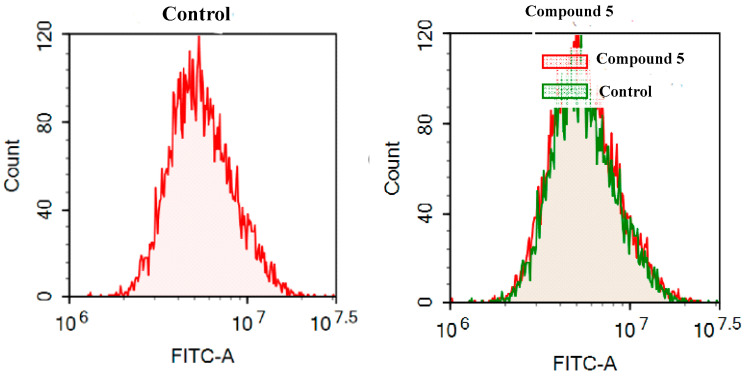
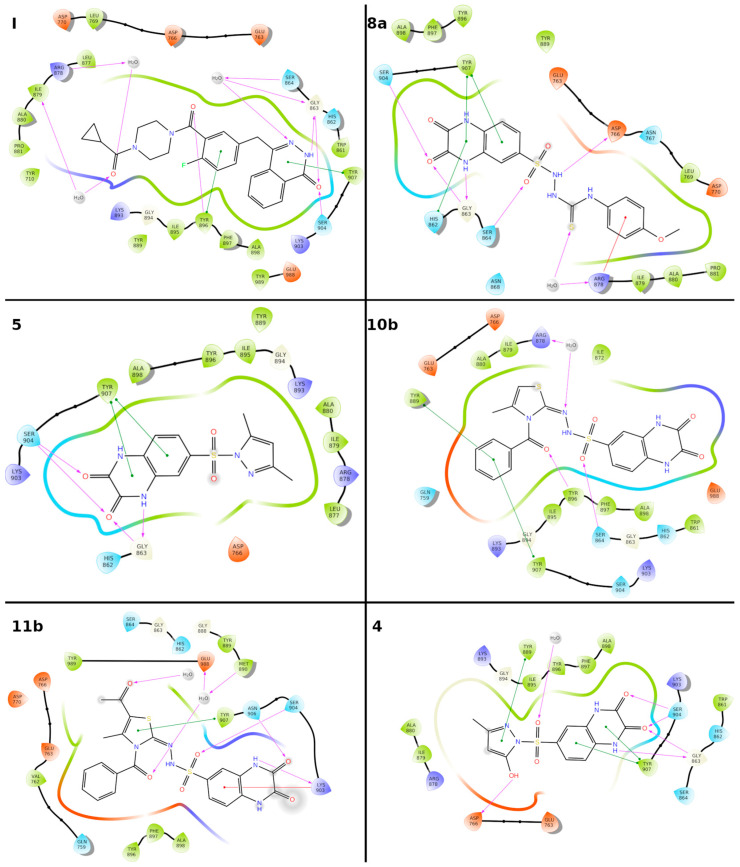
Herein, 2,3-dioxo-1,2,3,4-tetrahydroquinoxaline was used as a bio-isosteric scaffold to the phthalazinone motif of the standard drug Olaparib to design and synthesize new derivatives of potential PARP-1 inhibitory activity using the 6-sulfonohydrazide analog as the key intermediate. Although the new compounds represented the PARP-1 suppression impact of IC values in the nanomolar range, compounds , were the most promising suppressors, producing IC values of 2.31 and 3.05 nM compared to Olaparib with IC of 4.40 nM. Compounds , , and showed a mild decrease in the potency of the IC range of 6.35-8.73 nM. Furthermore, compounds , , , , and were evaluated as in vitro antiproliferative agents against the mutant BRCA1 (MDA-MB-436, breast cancer) compared to Olaparib as a positive control. Compound exhibited the most significant potency of IC; 2.57 µM, whereas the IC value of Olaparib was 8.90 µM. In addition, the examined derivatives displayed a promising safety profile against the normal WI-38 cell line. Cell cycle, apoptosis, and autophagy analyses were carried out in the MDA-MB-436 cell line for compound which exhibited cell growth arrest at the G2/M phase, in addition to induction of programmed apoptosis and an increase in the autophagic process. Molecular docking of the compounds , , , , and into the active site of PARP-1 was carried out to determine their modes of interaction. In addition, an in silico ADMET study was performed. The results evidenced that compound could serve as a new framework for discovering new potent anticancer agents targeting the PARP-1 enzyme.
在此,将 2,3-二氧代-1,2,3,4-四氢喹喔啉用作标准药物奥拉帕利的邻苯二甲酰亚胺基的生物等排体支架,以使用 6-磺酰基酰肼类似物 作为关键中间体设计和合成具有潜在 PARP-1 抑制活性的新衍生物。虽然新化合物代表了 PARP-1 抑制作用的 IC 值处于纳摩尔范围,但化合物 、 是最有前途的抑制剂,与 IC 值为 4.40 nM 的奥拉帕利相比,其 IC 值分别为 2.31 和 3.05 nM。化合物 、 、 的抑制作用稍弱,IC 值范围为 6.35-8.73 nM。此外,将化合物 、 、 、 、 作为体外抗增殖剂,与阳性对照奥拉帕利相比,对突变型 BRCA1(MDA-MB-436,乳腺癌)进行评估。化合物 表现出最显著的 IC 作用强度;2.57 µM,而奥拉帕利的 IC 值为 8.90 µM。此外,在所检查的衍生物中,对正常 WI-38 细胞系显示出有希望的安全性特征。在 MDA-MB-436 细胞系中对化合物 进行了细胞周期、细胞凋亡和自噬分析,结果表明该化合物可使细胞生长在 G2/M 期停滞,此外还可诱导程序性细胞凋亡和自噬过程增加。将化合物 、 、 、 分子对接到 PARP-1 的活性部位,以确定它们的相互作用方式。此外,还进行了计算机 ADMET 研究。结果表明,化合物 可作为一种新的框架,用于发现针对 PARP-1 酶的新型有效抗癌药物。