Section on Cellular Differentiation, Division of Translational Medicine, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20802, USA.
Istituto Giannina Gaslini, Largo Gaslini 5, 16147, Genoa, Italy.
Hum Mol Genet. 2023 Jan 6;32(2):262-275. doi: 10.1093/hmg/ddac197.
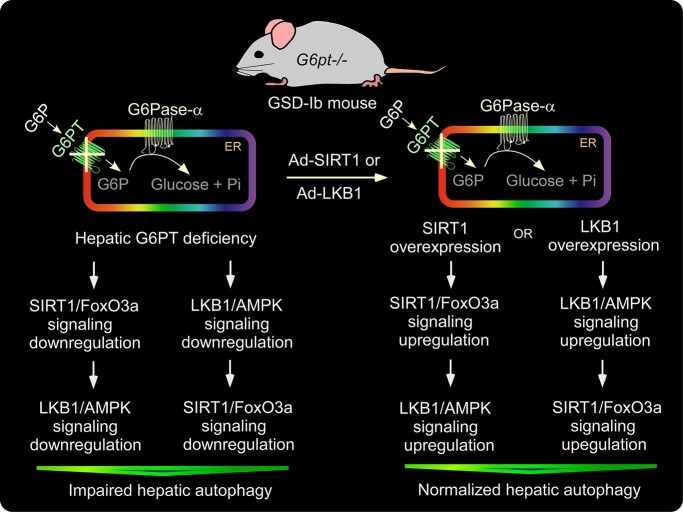
Type Ib glycogen storage disease (GSD-Ib) is caused by a deficiency in the glucose-6-phosphate (G6P) transporter (G6PT) that translocates G6P from the cytoplasm into the endoplasmic reticulum lumen, where the intraluminal G6P is hydrolyzed to glucose by glucose-6-phosphatase-α (G6Pase-α). Clinically, GSD-Ib patients manifest a metabolic phenotype of impaired blood glucose homeostasis and a long-term risk of hepatocellular adenoma/carcinoma (HCA/HCC). Studies have shown that autophagy deficiency contributes to hepatocarcinogenesis. In this study, we show that G6PT deficiency leads to impaired hepatic autophagy evident from attenuated expression of many components of the autophagy network, decreased autophagosome formation and reduced autophagy flux. The G6PT-deficient liver displayed impaired sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK) signaling, along with reduced expression of SIRT1, forkhead boxO3a (FoxO3a), liver kinase B-1 (LKB1) and the active p-AMPK. Importantly, we show that overexpression of either SIRT1 or LKB1 in G6PT-deficient liver restored autophagy and SIRT1/FoxO3a and LKB1/AMPK signaling. The hepatosteatosis in G6PT-deficient liver decreased SIRT1 expression. LKB1 overexpression reduced hepatic triglyceride levels, providing a potential link between LKB1/AMPK signaling upregulation and the increase in SIRT1 expression. In conclusion, downregulation of SIRT1/FoxO3a and LKB1/AMPK signaling underlies impaired hepatic autophagy which may contribute to HCA/HCC development in GSD-Ib. Understanding this mechanism may guide future therapies.
Ib 型糖原贮积病(GSD-Ib)是由葡萄糖-6-磷酸(G6P)转运蛋白(G6PT)缺乏引起的,该蛋白将 G6P 从细胞质转运到内质网腔,在那里腔内的 G6P 被葡萄糖-6-磷酸酶-α(G6Pase-α)水解为葡萄糖。临床上,GSD-Ib 患者表现出血糖稳态代谢表型受损和肝细胞腺瘤/癌(HCA/HCC)的长期风险。研究表明,自噬缺陷有助于肝癌的发生。在这项研究中,我们表明 G6PT 缺乏导致肝自噬受损,表现在自噬网络的许多成分表达减弱、自噬体形成减少和自噬流减少。G6PT 缺乏的肝脏表现出沉默信息调节因子 1(SIRT1)和 AMP 激活的蛋白激酶(AMPK)信号受损,同时 SIRT1、叉头框 O3a(FoxO3a)、肝激酶 B-1(LKB1)和活性 p-AMPK 的表达减少。重要的是,我们表明在 G6PT 缺乏的肝脏中过表达 SIRT1 或 LKB1 可恢复自噬和 SIRT1/FoxO3a 和 LKB1/AMPK 信号。G6PT 缺乏的肝脏中的脂肪变性降低了 SIRT1 的表达。LKB1 的过表达降低了肝甘油三酯水平,为 LKB1/AMPK 信号上调与 SIRT1 表达增加之间提供了潜在联系。总之,SIRT1/FoxO3a 和 LKB1/AMPK 信号的下调导致肝自噬受损,这可能导致 GSD-Ib 中 HCA/HCC 的发展。了解这种机制可能为未来的治疗提供指导。