Beniwal Meenu, Jain Neelam, Jain Sandeep, Aggarwal Navidha
Department of Pharmaceutical Education & Research, Bhagat Phool Singh Mahila Vishwavidyalaya, Khanpur Kalan, Sonepat, Haryana, 131301, India.
Department of Pharmaceutical Sciences, Guru Jambheshwar University of Science and Technology, Hisar, Haryana, 125001, India.
BMC Chem. 2022 Aug 17;16(1):61. doi: 10.1186/s13065-022-00852-8.
Aurora-A kinase is associated with the Aurora kinase family which has been considered a striking anticancer target for the treatment of human cancers.
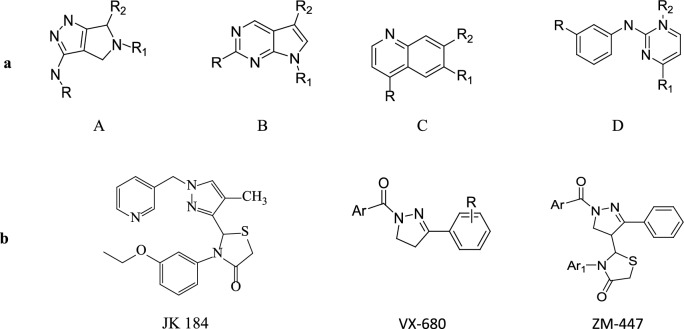
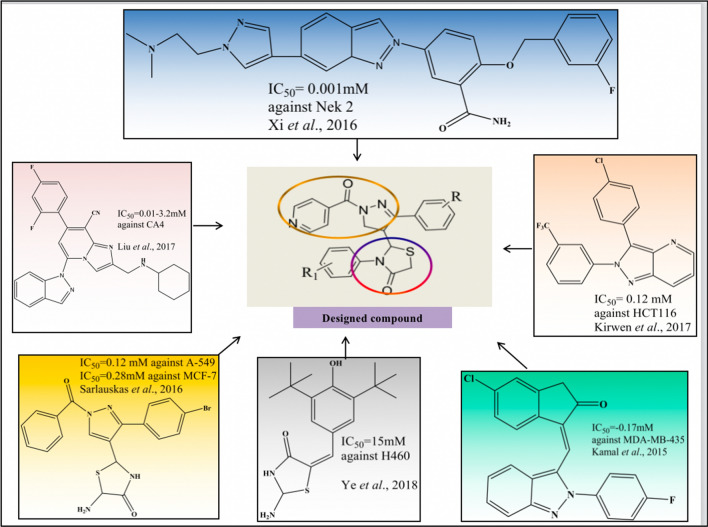
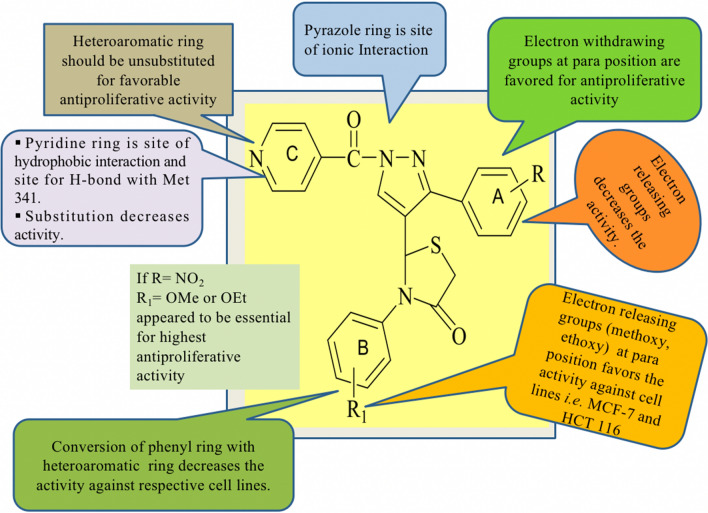
To design, synthesize, anticancer evaluation, and docking studies of novel 2-(1-isonicotinoyl-3-phenyl-1H-pyrazol-4-yl)-3-phenylthiazolidin-4-one derivatives as Aurora-A Kinase inhibitors.
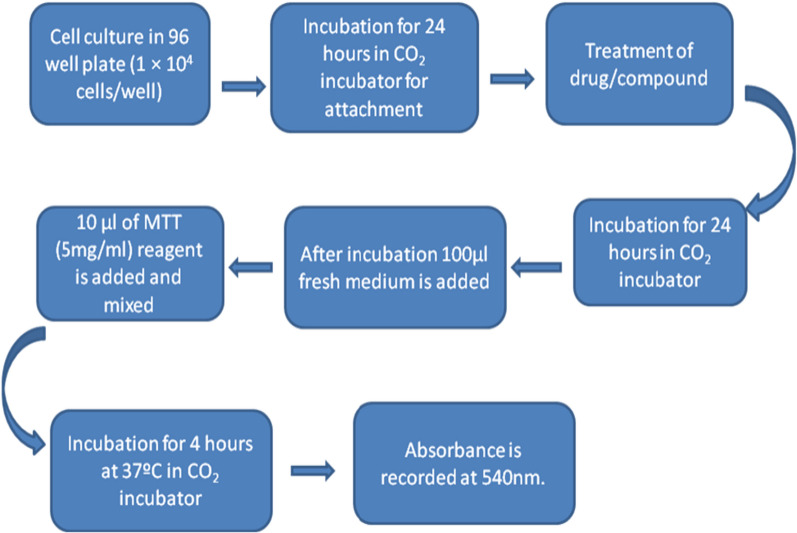
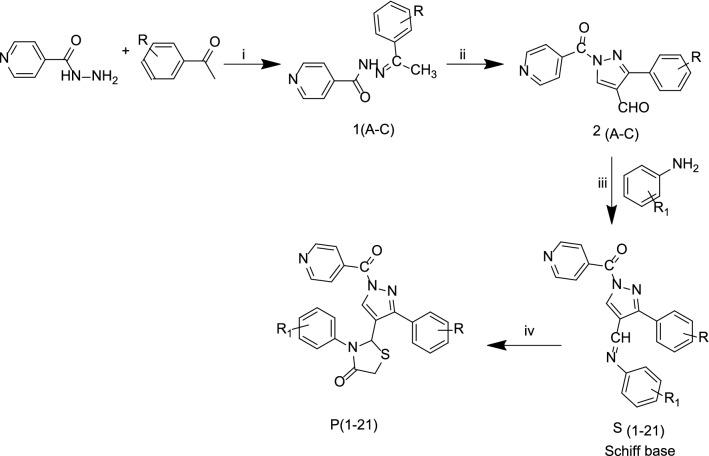
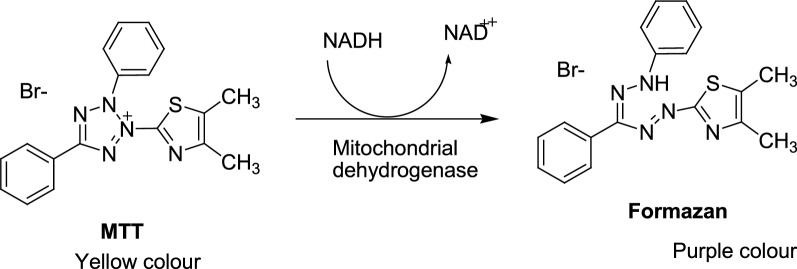
A total of 21 Pyrazole derivatives P (1-21) were synthesized by using the Vilsmeier Haack reagent which was characterized by FT-IR, H NMR, C NMR, and Mass spectroscopy. The synthesized derivatives were evaluated for their potential in vitro anticancer activity by MTT assay and Aurora-A kinase inhibition assay.
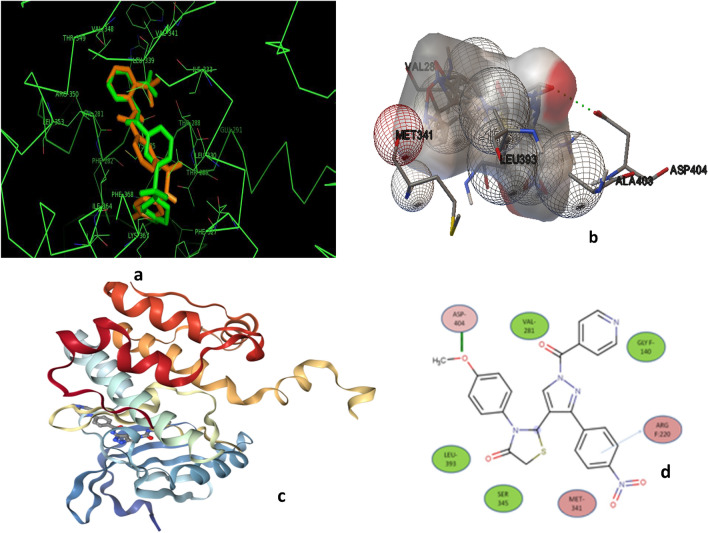
The cytotoxicity assay (MTT assay) showed that compound P-6 exhibited potent cytotoxicity (IC = 0.37-0.44 μM) against two cancer (HCT 116 and MCF-7) cell lines, which were comparable to the standard compound, VX-680. Compound P-6 also showed inhibition of Aurora-A kinase with an IC value of 0.11 ± 0.03 µM. A Docking study was done to compound P-6 and P-20 into the active site of Aurora A kinase, in order to get the probable binding model for further study.
A series of 21 novel pyrazole derivatives P(1-21) were designed, synthesized, in vitro anticancer evaluation, and docking studies for Aurora A kinase inhibition. The results established that P-6 is a prospective aspirant for the development of anticancer agents targeting Aurora-A kinase.
Aurora-A激酶与Aurora激酶家族相关,该家族被认为是治疗人类癌症的一个显著的抗癌靶点。
设计、合成新型2-(1-异烟酰基-3-苯基-1H-吡唑-4-基)-3-苯基噻唑烷-4-酮衍生物作为Aurora-A激酶抑制剂,并进行抗癌评估和对接研究。
使用Vilsmeier Haack试剂合成了总共21种吡唑衍生物P(1-21),通过傅里叶变换红外光谱(FT-IR)、氢核磁共振(H NMR)、碳核磁共振(C NMR)和质谱对其进行表征。通过MTT法和Aurora-A激酶抑制试验评估合成衍生物的体外抗癌活性潜力。
细胞毒性试验(MTT法)表明,化合物P-6对两种癌细胞系(HCT 116和MCF-7)表现出强效细胞毒性(IC = 0.37-0.44 μM),与标准化合物VX-680相当。化合物P-6还显示出对Aurora-A激酶的抑制作用,IC值为0.11±0.03 μM。对化合物P-6和P-20进行了对接研究,使其进入Aurora A激酶的活性位点,以获得可能的结合模型用于进一步研究。
设计、合成了一系列21种新型吡唑衍生物P(1-21),进行了体外抗癌评估和针对Aurora A激酶抑制的对接研究。结果表明,P-6是开发靶向Aurora-A激酶抗癌药物的一个有前景的候选物。