Faculty of Engineering and Natural Sciences, Sabanci University, 34956Istanbul, Turkey.
J Chem Inf Model. 2022 Dec 26;62(24):6715-6726. doi: 10.1021/acs.jcim.2c00507. Epub 2022 Aug 19.
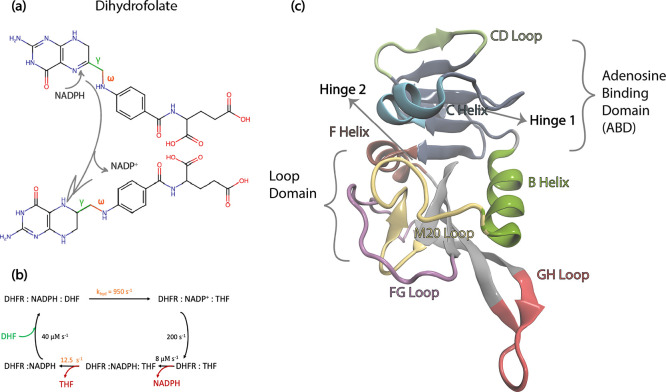
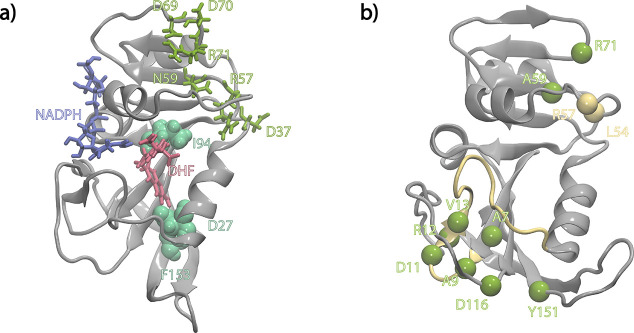
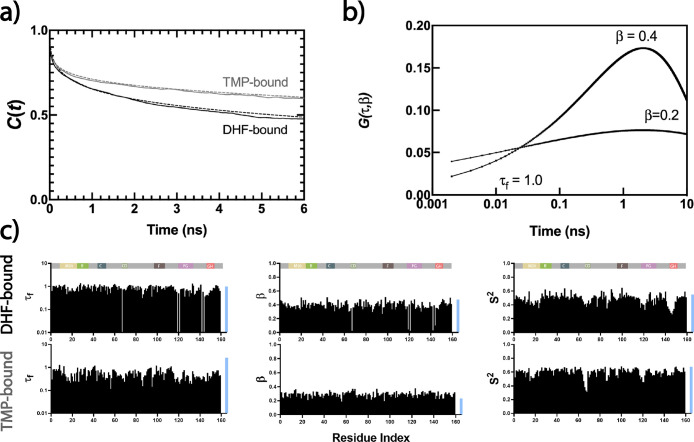
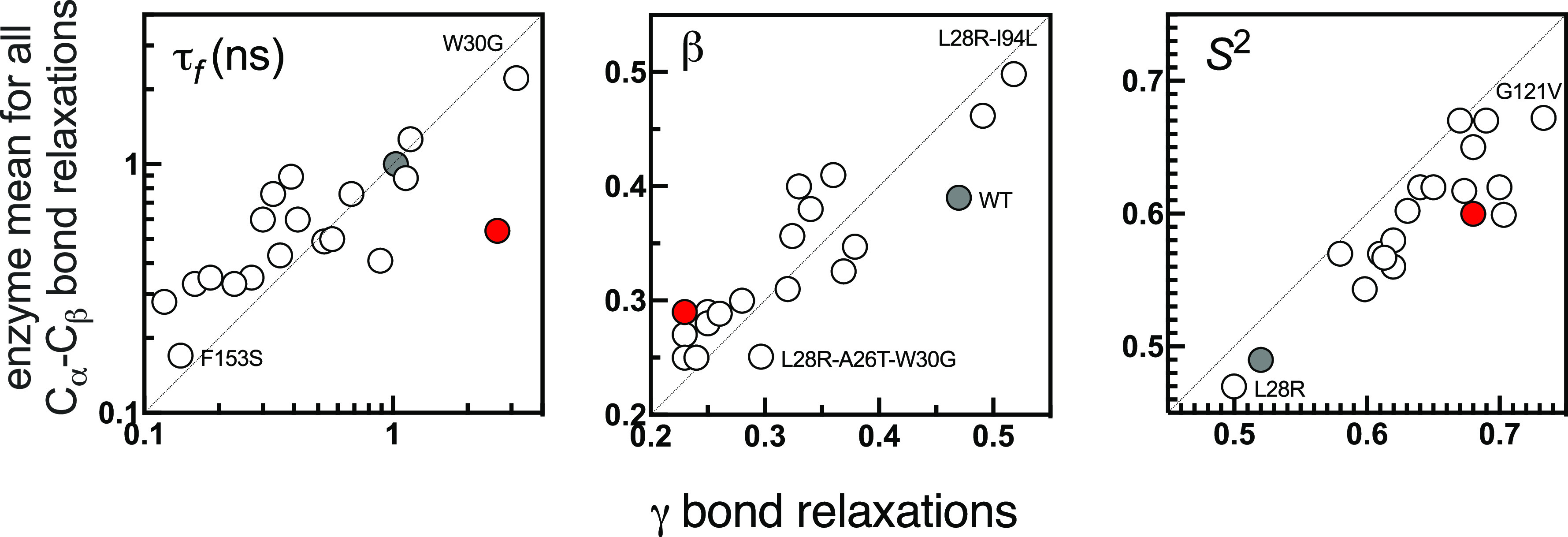
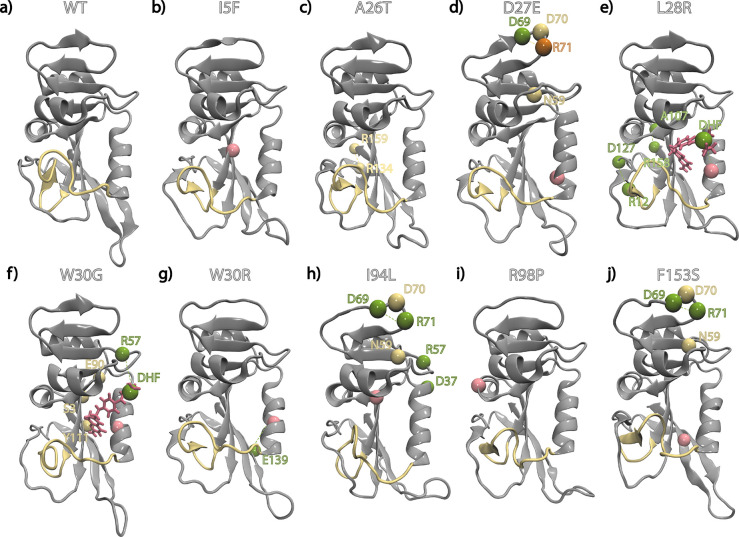
Antibiotic resistance is a global health problem in which mutations occurring in functional proteins render drugs ineffective. The working mechanisms of the arising mutants are seldom apparent; a methodology to decipher these mechanisms systematically would render devising therapies to control the arising mutational pathways possible. Here we utilize C-C bond vector relaxations obtained from moderate length MD trajectories to determine conduits for functionality of the resistance conferring mutants of dihydrofolate reductase. We find that the whole enzyme is synchronized to the motions of the substrate, irrespective of the mutation introducing gain-of-function or loss-of function. The total coordination of the motions suggests changes in the hydrogen bond dynamics with respect to the wild type as a possible route to determine and classify the mode-of-action of individual mutants. As a result, nine trimethoprim-resistant point mutations arising frequently in evolution experiments are categorized. One group of mutants that display the largest occurrence (L28R, W30G) work directly by modifying the dihydrofolate binding region. Conversely, W30R works indirectly by the formation of the E139-R30 salt bridge which releases energy resulting from tight binding by distorting the binding cavity. A third group (D27E, F153S, I94L) arising as single, resistance invoking mutants in evolution experiment trajectories allosterically and dynamically affects a hydrogen bonding motif formed at residues 59-69-71 which in turn modifies the binding site dynamics. The final group (I5F, A26T, R98P) consists of those mutants that have properties most similar to the wild type; these only appear after one of the other mutants is fixed on the protein structure and therefore display clear epistasis. Thus, we show that the binding event is governed by the entire enzyme dynamics while the binding site residues play gating roles. The adjustments made in the total enzyme in response to point mutations are what make quantifying and pinpointing their effect a hard problem. Here, we show that hydrogen bond dynamics recorded on sub-μs time scales provide the necessary fingerprints to decipher the various mechanisms at play.
抗生素耐药性是一个全球性的健康问题,其中功能蛋白的突变会使药物失效。产生的突变体的工作机制很少是明显的;一种系统地破译这些机制的方法将有可能设计出控制新出现的突变途径的疗法。在这里,我们利用从中等长度 MD 轨迹中获得的 C-C 键向量松弛来确定二氢叶酸还原酶耐药突变体的功能途径。我们发现,整个酶与底物的运动同步,无论突变是引入功能获得还是功能丧失。运动的总协调表明,与野生型相比,氢键动力学的变化可能是确定和分类单个突变体作用模式的一种途径。结果,对在进化实验中经常出现的 9 个甲氧苄啶耐药点突变进行了分类。一组显示最大发生率的突变体(L28R、W30G)直接通过修饰二氢叶酸结合区域起作用。相反,W30R 通过形成 E139-R30 盐桥起作用,该盐桥释放由于紧密结合而产生的能量,从而通过扭曲结合腔来改变结合腔。第三组(D27E、F153S、I94L)在进化实验轨迹中作为单个、引起耐药性的突变体出现,它们变构和动态地影响在残基 59-69-71 形成的氢键基序,进而修饰结合位点动力学。最后一组(I5F、A26T、R98P)由与野生型最相似的性质的突变体组成;这些突变体仅在另一个突变体固定在蛋白质结构上后才出现,因此显示出明显的上位性。因此,我们表明结合事件由整个酶动力学控制,而结合位点残基起门控作用。对整个酶的调整以响应点突变是使量化和精确定位它们的效果成为一个难题的原因。在这里,我们表明在亚微秒时间尺度上记录的氢键动力学提供了破译各种起作用机制的必要指纹。